
ABSTRACT
Though DNMTs inhibitors were widely used in myelodysplastic syndrome and leukaemia, their application in solid tumours has been limited by low response rate and lack of optimal combination strategies. In gastric cancer (GC), the therapeutic implication of KRAS mutation or MEK/ERK activation for combinational use of DNMTs inhibitors with MEK/ERK inhibitors remains elusive. In this study, stable knockdown of DNMT1 expression by lentiviral transfection led to decreased sensitivity of GC cells to 5-Azacytidine. KRAS knockdown in KRAS mutant GC cells or the MEK/ERK activation by EGF stimulation in GC cells increased DNMT1 expression, while inhibition of MEK/ERK activity by Selumetinib led to decreased DNMT1 expression. 5-Azacytidine treatment, which led to dramatic decline of DNMTs protein levels and increased activity of MEK/ERK pathway, altered the activity of MEK/ERK inhibitor Selumetinib on GC cells. Both RAS-dependent gene expression signature and expression levels of multiple MEK/ERK-dependent genes were correlated with DNMT1 expression in TCGA stomach cancer samples. In conclusion, DNMT1 expression partially dictates 5-Azacytidine sensitivity and correlates with RAS/MEK/ERK activity in GC cells. Combining DNMTs inhibitor with MEK/ERK inhibitor might be a promising strategy for patients with GC.
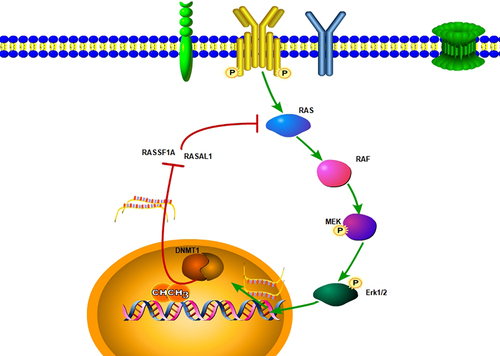
Introduction
Epigenetic alterations such as DNA hyper-methylation, which lead to chromatin remodelling and dysregulation of cancer-related genes, are essential for cancer development [Citation1,Citation2]. DNA methyltransferases (DNMTs), responsible for DNA hyper-methylation in cancer development, are the enzymes that add methyl groups to the 5 position of cytosine residues of CpG dinucleotides [Citation3]. Reversal of DNA hyper-methylation by DNMTs inhibitors is becoming an important treatment option for patients with cancer [Citation4]. Though DNMTs inhibitors were widely used in myelodysplastic syndrome and leukaemia, their application in solid tumours has been limited by low response rate and lack of optimal combination strategies [Citation5]. Therefore, it is of critical importance to identify biomarkers to predict sensitivity to DNMTs inhibitors and develop combinational strategy for their clinical application in solid tumours [Citation4].
DNA methylation is catalysed by three main DNA methyltransferases (DNMTs): DNA methyltransferase 1 (DNMT1), DNA methyltransferase 3A (DNMT3A), and DNA methyltransferase 3B (DNMT3B) [Citation3]. Among the DNA methyltransferases, DNMT1 is thought to be the predominant maintenance methyltransferase gene [Citation3,Citation6]. Silencing of numerous tumour suppressor genes, mediated by DNMT1 methyltransferase, is crucial for the carcinogenesis and progression of GC [Citation7,Citation8]. In the meantime, the potential of targeting DNMT1 as a therapeutic option for patients GC, was proven in various studies [Citation9,Citation10].
The Ras/MEK/ERK pathway mediates various cellular processes, which include cell growth, proliferation, and differentiation [Citation11]. Aberrant activation of RAS/MEK/ERK pathway occurs in more than 30% of human cancers and has been documented as a driving force of proliferation and carcinogenesis for various malignancies [Citation12,Citation13]. Though less frequently detected compared with other malignancies like pancreatic cancer, KRAS mutation or amplification was detectable and indicated poor prognosis in patients with GC [Citation14–16]. In GC, the therapeutic implication of KRAS mutation or MEK/ERK activation for combinational use of DNMTs inhibitors with MEK/ERK inhibitors remains elusive. Moreover, the correlation between MEK/ERK pathway activity and DNMT1 expression has not yet been fully elucidated in GC.
In this study, we first assessed GC cell sensitivity to 5-Azacytidine and explored the role of DNNT1 in conferring sensitivity to DNMTs inhibition. Then, the correlation between DNMT1 expression and RAS/MEK/ERK pathway activity and the prospect of utilizing 5-Azacytidine for combinational treatment with MEK/ERK inhibitor Selumetinib in GC cells were explored.
Material and methods
Cell lines
The human GC cell lines were maintained in our lab and routinely cultured in RPMI-1640 medium (GIBCO, Thermo Fisher Scientific, Waltham, MA, USA), containing with 10% fetal bovine serum (GIBCO, Thermo Fisher Scientific, Waltham, MA, USA), 100 mg/ml streptomycin, and 100 U/ml penicillin sodium. Moreover, the STR profiles were detected and matched with ATCC fingerprints (ATCC. org) before using these cell lines.
Antibody and reagents
5-Azacytidine and CCK-8 were purchased from GLPBIO (Montclair, USA). MEK/ERK inhibitor Selumetinib was purchased from MedChemExpress (Monmouth Junction, NJ, USA). ECL substrate was purchased from 4A Biotech Co. Ltd (Beijing, China). KRAS antibody was purchased from Abcam (Cambridge, UK). α-Tubulin antibody was purchased from Proteintech (Rosemont, IL, USA). DNMT1, P44/42 MAPK (Erk1/2), phospho-P44/42 MAPK (p-Erk1/2) (Thr202/Tyr204), MEK1/2, and phospho-MEK1/2 (Ser217/221) antibodies were purchased from CST (Danvers, MA, USA).
RT-PCR
Total RNAs were isolated using Trizol reagent (Takara, Otsu, Shiga, Japan)) according to the manufacturer’s instructions. First-strand cDNA was reversely transcribed from 0.1 µg total RNA in a final volume of 10 µl using reverse transcription reagent kit from Takara (Otsu, Shiga, Japan) according to the manufacturer’s instructions. PCR reaction was performed with with a reagent kit from Takara (Otsu, Shiga, Japan) in a PCR cycler (Thermal) according to a protocol as follows: 1 cycle of 95°C for 30 s; 45 cycles of 95°C for 10 s, extension 60°C for 20 s; and holding at 4°C. The 2−∆∆Ct method was used to calculate relative RNA expression. The primer sequences are listed in Supplementary Table S1.
Western blotting
Cells were lysed in RIPA lysis buffer (Beyotime Biotechnology, Shanghai, China) supplemented with a protease inhibitor cocktail (Roche, Germany) and a phosphatase inhibitor cocktail (Roche, Germany). Proteins from lysed cells were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a nitrocellulose membrane. Nonspecific binding was then blocked for 2 hours at room temperature in phosphate buffer containing 5% non-fat milk. Membranes were incubated overnight at 4°C with specific primary antibodies. DNMT1 (1:1000, CST, #28754), KRAS (1:1000, Abcam, ab275876), P44/42 MAPK (Erk1/2) (1:1000, CST, #4695), phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (1:1000, CST, #4370), MEK1/2 (1:1000, CST, #8727), phospho-MEK1/2 (Ser217/221) (1:1000, CST, #9154), and α-Tubulin (1:20000, Proteintech, 6603–1-Ig).
SiRNA transfection
SiRNAs were designed and synthesized from RiboBio (Guangzhou, China) or Sangon Biotech (Shanghai, China). The negative control siRNA has no significant sequence similarity to mouse, rat, or human gene sequence. The target sequence information was listed in Supplementary Table S2. For siRNA effect validation, a density of 200,000 per well was plated in 6-well plates. The following day, a stock solution containing 6 μl (20 μmol/ml) of siRNA and 6 μl of transfection reagent per 1000 μl Opti-MEM (Thermal Fisher Technologies) was prepared and incubated at room temperature for 15 min. Then, the medium was aspirated from each well and replaced with 1000ul Opti-MEM containing siRNA transfection mix.
Cells exhibiting stable down-regulation of DNMT1 expression were established by transduction with lentivirus (hU6-MCS-Ubiquitin-EGFP-IRES-puromycin) expressing DNMT1 shRNA: LV-shDNMT1 (#1, target sequence: GCCCAATGAGACTGACATCAA) (Genetech, Shanghai, China). Control cells were transfected with negative control virus CON313 (LV-shControl, target sequence: TTCTCCGAACGTGTCACGT). HiTransG A and HiTransG P were used as transfection agents. Cells infected with lentivirus were selected with puromycin for 2 weeks.
Chemo-sensitivity analysis
Chemo-sensitivity analyses for 5-Azacytidine or/and Selumetinib were performed by measuring the metabolic activity of the cells using the CCK-8 assay, which was used as a surrogate for cell viability. After incubating for 48-96 h, the cells were then incubated with a CCK-8 solution (GLPBIO, USA) for 2 hours. The absorbance at 450 nm was measured with a Varioskan Flash spectrometer (Thermo Fisher Scientific, Waltham, USA). The IC50, the half maximal inhibitory concentration, was determined by interpolation of dose-response curves. For combined treatment of Selumetinib and 5-Azacytidine, the application schedule with clinically relevant concentrations of both agents was used. 5-Azacytidine: 1uM or 10uM for 24 h followed by Selumetinib (1uM or 10uM) and 5-Azacytidine combined incubation for another 48-72 h. To assess the effects of combined treatment of Selumetinib and 5-Azacytidine, both CCK-8 assay and LDH assay were used. For the LDH assay, after incubation in 96-well plate the cells were then incubated with Lysis Solution, followed by Working Solution and Stop Solution (GLPBIO, USA). Then, to measure the total LDH release, the absorbance at 490 nm was measured with a Varioskan Flash spectrometer (Thermo Fisher Scientific, Waltham, USA). The effect of drugs on cell proliferation was calculated as percentage growth of cells relative to respective controls (DMSO-treated cells). CompuSyn software (Cambridge, UK) was used to calculate combination index. Combination index (CI) values less than 1.0 indicate a synergistic interaction of the two agents in the combination.
Immunohistochemistry (IHC)
IHC was performed on sections of tumour tissue samples from GC patients as previously described [Citation17]. The sections were incubated with the diluted antibody: DNMT1 (ab188453, 1:100). The final immunoactivity score of each section was determined by two independent observers in a blinded manner. IHC scores of 0–12 were calculated and graded as negative (−, score: 0), weak (+, score: 1–4), moderate (+ +, score: 5–8), or strong (+ + +, score: 9–12).
Statistical analysis
Data are expressed as mean ± standard deviation (SD). The Student’s t-test or Fisher’s exact test was used to compare differences between the two experimental groups. Differences were considered significant when p < 0.05.
Results
The DNMT1 expression level partially dictates 5-Azacytidine sensitivity in GC cells
We first detected the expression level of DNMT1 in a subset of GC cells and immortalized gastric epithelial GES-1 cells by western blotting and RT-PCR, respectively (). Then, the intrinsic sensitivity to 5-Azacytidine was determined by CCK-8 assay in the above cells. The results showed that MKN-45, MKN-28, SNU-1, and GES-1 cells were relatively sensitive to 5-Azacytidine treatment (IC50 < 4uM), while AGS, BGC-823, SGC-7901, and MGC-803 cells were relatively resistant (IC50 > 4uM) ().
Figure 1. The DNMT1 expression level partially dictates 5-Azacytidine sensitivity in GC cells.
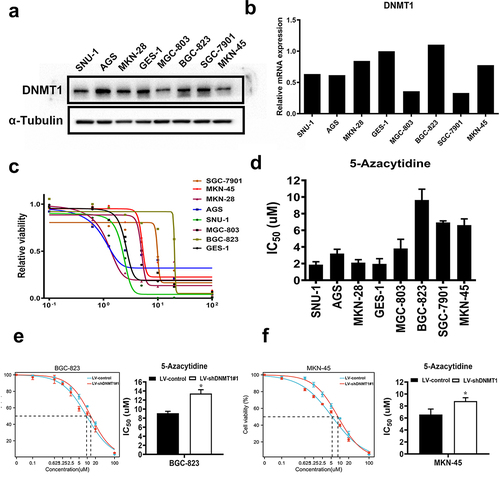
We then determined whether DNMT1 expression dictates 5-Azacytidine sensitivity in GC cells. DNMT1 was stably knocked down by lentiviral transfection, and the efficiency of DNMT1 knockdown was validated by western blotting in 5-Azacytidine relatively resistant BGC-823 and relatively sensitive MKN-45 cells (Supplementary Figure S1). IC50 value to 5-Azacytidine was significantly increased upon DNMT1 knockdown in BGC-823 and MKN-45 cells ().
MEK/ERK inhibition led to decreased DNMT1 expression in GC cells
Previous study showed that RAS/MEK/ERK activity promoted DNA methylation by increasing DNMT1 expression in colon cancer cells [Citation18]. Therefore, we speculated a similar phenomenon might exist in GC cells. First, we determined the activity of MEK/ERK pathway in GC cells by detecting the p-Erk1/2 level by western blotting (Supplementary Figure S2). In MEK/ERK moderately hyperactive BGC-823 and highly hyperactive AGS cells, Selumetinib treatment significantly inhibited the activity of MEK/ERK pathway (indicated by p-Erk1/2 level) and led to decreased DNMT1 expression (). EGF stimulation, which induced significant activation of MEK/ERK pathway, also led to increased expression of DNMT1 in both MEK/ERK moderately hyperactive BGC-823 and MEK/ERK hypoactive MKN-45 cells (Supplementary Fig. S3, ).
Figure 2. MEK/ERK inhibition led to decreased DNMT1 expression in GC cells.
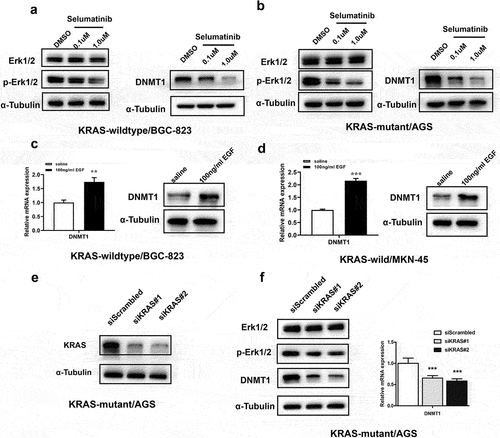
In KRAS mutant and MEK/ERK hyperactive AGS cells, KRAS was knocked down by siRNA transfection (). Significant decrease of MEK/ERK pathway activity and concurrent down-regulation of DNMT1 expression were observed upon KRAS knockdown in AGS cells ().
5-Azacytidine treatment led to significant decline of DNMT1 and DNMT3A protein levels in GC cells
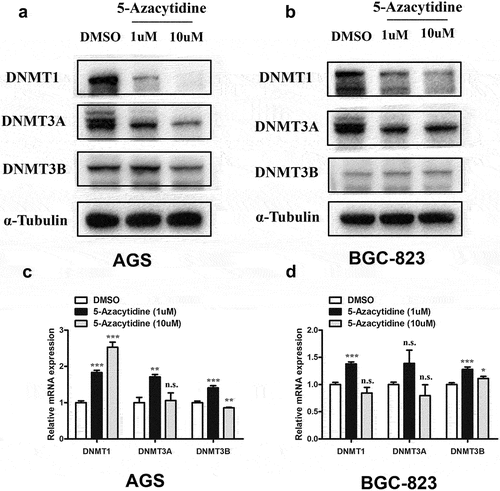
It was observed that 5-Azacytidine treatment led to dose-dependent down-regulation of DNMT1 protein expression in both KRAS mutant AGS and KRAS wild-type BGC-823 cells (). Similarly, dramatic decline in protein expression level of DNMT3A was also observed in both AGS and BGC-823 cells upon 5-Azacytidine treatment (). A decline of DNMT3B protein level was observed upon high dosage (10uM) of 5-Azacytidine treatment in AGS cells (). RT-PCR results showed that 1uM 5-Azacytidine treatment led to increased mRNA levels of DNMT1, DNMT3A, and DNMT3B (). However, high concentration of 5-Azacytidine (10uM) treatment led to inconsistent alteration of the DNMT1 and DNMT3B mRNA levels between AGS and BGC-823 cells, while no significant effect on DNMT3A mRNA levels was observed ().
Figure 3. 5-Azacytidine treatment led to significant decline of DNMT1 and DNMT3A protein levels in GC cells.
5-Azacytidine pre-treatment led to increased activity of the MEK/ERK pathway in GC cells
We also observed that 5-Azacytidine treatment led to the increased activity of MEK/ERK pathway, as indicated by the phosphorylation levels of MEK1/2 and ERK1/2 (p-MEK1/2 and p-Erk1/2) in KRAS wild-type BGC-823 and KRAS mutant AGS cells (). As the activity of the MEK/ERK pathway was correlated with the sensitivity to MEK/ERK inhibitors, we set out to explore whether 5-Azacytidine pre-treatment altered the activity of Selumetinib on GC cells. Interestingly, a synergistic effect (CI value < 1) was observed when a relatively high concentration of Selumetinib (10uM) was combined with high concentration of 5-Azacytidine (10uM) in BGC-823 cells (, ). Moreover, in KRAS mutant and MEK/ERK hyperactive AGS cells, both high-concentration and low-concentration combination of 5-Azacytidine and Selumetinib produced strong synergistic effects (CI value < 0.8, and ).
Figure 4. 5-Azacytidine pre-treatment led to increased activity of the MEK/ERK pathway in GC cells.
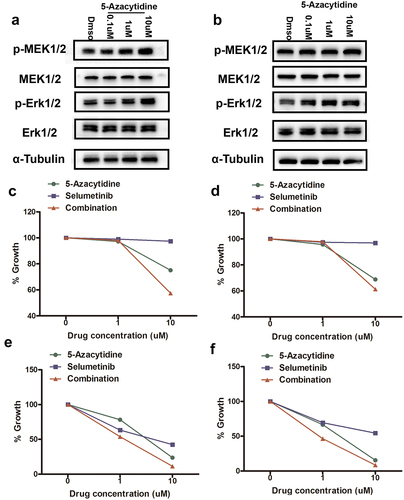
Table 1. Combinational index (CI) value for sequential treatment with 5-Azacytidine and Selumetinib in AGS and BGC-823 cells. CI value: <1 synergistic; = 1 Additive; and > 1 Antagonistic.
DNMT1 expression was correlated with RAS/MEK/ERK activity in GC
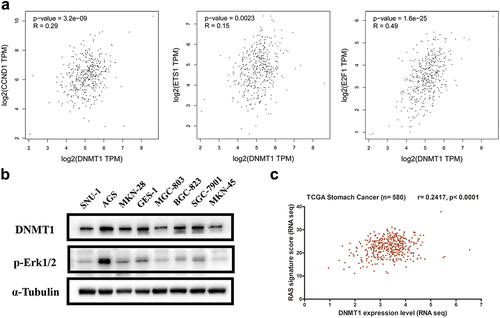
To determine a possible correlation of DNMT1 expression with RAS/MEK/ERK activity in GC, public database containing gene expression data of TCGA GC samples was accessed. Analysis revealed that among the genes showing highly significant correlations with DNMT1 expression in TCGA GC samples, numerous MEK/ERK pathway-dependent genes were enriched, which included ETS-1, E2F1, and CCND1 (). Western blotting analysis showed that MEK/ERK pathway activity, indicated by p-Erk1/2 level, was higher in DNMT1 relatively high-expressing AGS, GES-1, SGC-7901, and BGC-823 cells (). Furthermore, the DNMT1 expression level was also significantly correlated with RAS-dependent gene signature (r = 0.2417, p < 0.0001) ().
Figure 5. DNMT1 expression was correlated with RAS/MEK/ERK activity in GC.
DNMT1 was up-regulated in GC tumour tissues compared with adjacent normal tissues
Then, we detected the DNMT1 expression by immunohistochemistry in a panel of 20 GC tissues and observed an increased expression of DNMT1 in majority of tumour tissues compared with paired adjacent normal tissues ().
Figure 6. DNMT1 was up-regulated in GC tumour tissues compared with adjacent normal tissues.
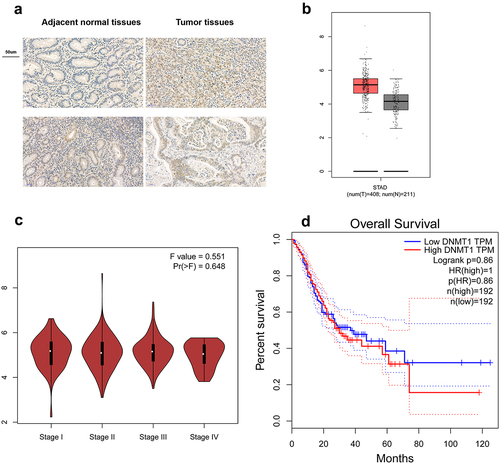
Bioinformatics analysis of TCGA GC database revealed that DNMT1 was significantly higher in GC tumour tissues than normal tissues (). However, DNMT1 expression was not increased in high-stage (III/IV) GC tumour tissues compared with low-stage (I/II) GC tumours tissues (). Moreover, DNMT1 expression was not significantly correlated with the prognosis of GC patients ().
Discussion
5-Azacytidine and 5-Aza-deoxycytidine (decitabine) are both chemical analogs of cytidine and important DNMTs inhibitors approved by the US FDA for myelodysplastic syndrome leukaemia and leukaemia treatment [Citation19,Citation20]. In more recent years, the immunomodulatory and anti-tumour effects of DNMTs inhibitors were described in solid tumours [Citation21–24]. However, the application of DNMTs inhibitors in solid tumours is challenged by low response rate and lack of optimal combination strategies [Citation24,Citation25]. Therefore, the discovery and identification of biomarkers to predict response are urgently needed. Meanwhile, the development of alternative treatment options such as optimal combination strategy is imperative.
Jia Yu et al showed that 5-Aza-deoxycytidine (decitabine) sensitivity in 45 solid tumour cells was associated with DNMT1 mRNA level [Citation26]. Another study showed that high DNMT3B protein level dedicated 5-Aza-deoxycytidine hypersensitivity in Testicular Germ Cell Tumors [Citation27]. The results from organoid culture originating from triple negative breast cancer PDX models showed that 5-Aza-deoxycytidine sensitivity was highly correlated with both DNMT3A and DNMT3B protein levels [Citation28]. In this study, we observed that DNMT1 expression partially dictated 5-Azacytidine sensitivity, as shown by increased IC50 to 5-Azacytidine in DNMT1 knockdown GC cells. Likely due to the small number of GC cell lines detected, we did not observe a significant correlation between DNMT1 expression and 5-Azacytidine sensitivity (data not shown). It is suggested that the difference in mode of action among different DNMTs inhibitors, and tumour heterogeneity might be the factors associated with the discrepancy of the above results [Citation29,Citation30]. Moreover, the pre-clinical models, which more faithfully represent human tumour biology and clinical treatment response, are warranted to discover or validate the predictive value of these biomarkers. Our results further showed 5-Azacytidine treatment led to dramatic decline of DNMT1 and DNMT3A protein levels in GC cells, which suggests that 5-Azacytidine might mainly function through DNA methyltransferase DNMT1 or DNMT3A in GC cells.
Previous study showed that 5-Aza-deoxycytidine treatment induced significant activation of MEK/ERK pathway in ovarian cancer cells [Citation26]. We observed that treatment with 5-Azacytidine also increased the activity of the MEK/ERK pathway in both KRAS wild-type and KRAS-mutant GC cells. Given the fact that the increased activity of MEK/ERK correlated with the sensitivity to MEK/ERK inhibitors in tumour cells, we further explored the effect of combining MEK/ERK inhibitor Selumetinib with 5-Azacytidine. Interestingly, when Selumetinib of relatively high concentration (10uM) was combined with 5-Azacytidine of approximately IC50 value (10uM), a synergistic effect was observed in KRAS wild-type BGC-823 cells. More importantly, strong synergistic effects were also observed in KRAS mutant and MEK/ERK hyperactive GC AGS cells, irrespective of the drug concentration. These results suggested that the activation status of RAS/MER/ERK pathway and the combination dosage might be the key factors determining the effects of the combined treatment.
RAS/MEK/ERK signalling was reported to regulate DNMT1 expression and DNMT1-dependent DNA methylation in various tumours [Citation18,Citation31,Citation32]. In this study, EGF-induced MEK/ERK activation led to the increased DNMT1 expression in GC cells. Moreover, decrease of MEK/ERK activity by KRAS siRNA transfection in KRAS mutant GC cells or small-molecule inhibitor of MEK/ERK pathway in MEK/ERK hyperactive GC cells decreased DNMT1 expression. On the one hand, silencing of tumour suppressor genes by DNMT1 methyltransferase activity is essential for oncogenic transformation by genetic mutation like RAS activation mutation [Citation33,Citation34]. On the other hand, a combination of genetic mechanisms with epigenetic alterations might account for the oncogenic transformation in various tumours [Citation35,Citation36].
Then, both immunohistochemistry and bioinformatics analysis showed that DNMT1 expression is increased in GC tumour tissues compared with adjacent normal tissues, which is consistent with previous findings that DNMT1 is expressed at higher levels in early gastric cancer or high-grade intraepithelial neoplasia tissues compared to that in non-cancer mucosa or low-grade intraepithelial neoplasia tissues [Citation37]. Mechanistically, H. pylori-induced chronic inflammation, but not H. Pylori itself, played a direct role in the induction of DNMT1 expression and aberrant DNA methylation in GC [Citation38]. Moreover, suppression of aberrant DNA methylation by a demethylating agent was shown to inhibit gastric cancer development in an animal model [Citation39]. However, DNMT1 expression was found not correlated with higher tumour stage and poorer prognosis in patients with GC. Taken together, we proposed that DNMT1 is more likely to play critical roles in the initial stages of GC, but not in the advanced stages of GC.
The interaction between DNMT1 and RAS/MEK/ERK pathway has been proven to be one key molecular cascade in tumour carcinogenesis [Citation34,Citation35,Citation40,Citation41]. Prior studies have shown that via inactivation of RAS inhibitor genes, which included DAB2, RASSF1A, RASAL1, and DAB2IP, DNA hyper-methylation was associated with aberrant activation of RAS signalling and increased tumorigenesis [Citation35,Citation41,Citation42]. We also observed the increased expression of RASSF1A, RASAL1, and DAB2IP upon DNMT1 knockdown or DNMTs inhibition by 5-Azacytidine in GC cells (data not shown). Whether and how the expression or activity of DNMT1 might affect the activity of RAS/MEK/ERK pathway in GC warrant further investigation. Moreover, the molecular mechanisms underlying the interaction between DNMT1 methyltransferase and RAS/MEK/ERK pathway in GC remain to be elucidated.
In summary, the RAS/MEK/ERK-DNMT1 signalling pathway in GC was validated in our study. Our results also suggested that combined targeting of DNA methyltransferases with MEK/ERK inhibitors might be a promising treatment option for patients with GC. Future studies are needed to test the therapeutic benefit of this strategy in animal models and ultimately in patients with GC.
Author contribution
Zhangqian Chen, Lin Zhang, and Yang Yang contributed to acquisition of data, analysis and interpretation of data, statistical analysis, and writing original draft; Haiming Liu and Xiaoyu Kang contributed to software and data curation; Daiming Fan, Yongzhan Nie, and Zhangqian Chen contributed to study concept and design, reviewing and editing of the manuscript, and study supervision.
Availability of data
The data that support the findings of this study are available from the corresponding author, Daiming Fan, upon reasonable request.
Supplemental Material
Download Zip (9.6 MB)Acknowledgments
We appreciated the technical support from Jing Wang and Jiawen Wei (Junior laboratory technician, Xijing Hospital, Fourth Military Medical University).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15592294.2023.2254976
Additional information
Funding
References
- Zhao S, Allis CD, Wang GG. The language of chromatin modification in human cancers. Nat Rev Cancer. 2021;21(7):413–13. doi: 10.1038/s41568-021-00357-x
- Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128(4):683–692. doi: 10.1016/j.cell.2007.01.029
- Lyko F. The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat Rev Genet. 2018;19(2):81–92. doi: 10.1038/nrg.2017.80
- Gnyszka A, Jastrzebski Z, Flis S. DNA methyltransferase inhibitors and their emerging role in epigenetic therapy of cancer. Anticancer Res. 2013;33:2989–2996.
- Cowan LA, Talwar S, Yang AS. Will DNA methylation inhibitors work in solid tumors? A review of the clinical experience with azacitidine and decitabine in solid tumors. Epigenomics. 2010;2(1):71–86. doi: 10.2217/epi.09.44
- Song J, Teplova M, Ishibe-Murakami S, et al. Structure-based mechanistic insights into DNMT1-mediated maintenance DNA methylation. Science. 2012;335(6069):709–712. doi: 10.1126/science.1214453
- Hino R, Uozaki H, Murakami N, et al. Activation of DNA methyltransferase 1 by EBV latent membrane protein 2A leads to promoter hypermethylation of PTEN gene in gastric carcinoma. Cancer Res. 2009;69:2766–2774. doi: 10.1158/0008-5472.CAN-08-3070
- Chen W, Gao N, Shen Y, et al. Hypermethylation downregulates Runx3 gene expression and its restoration suppresses gastric epithelial cell growth by inducing p27 and caspase3 in human gastric cancer. J Gastroenterol Hepatol. 2010;25(4):823–831. doi: 10.1111/j.1440-1746.2009.06191.x
- Mutze K, Langer R, Schumacher F, et al. DNA methyltransferase 1 as a predictive biomarker and potential therapeutic target for chemotherapy in gastric cancer. Eur J Cancer. 2011;47:1817–1825. doi: 10.1016/j.ejca.2011.02.024
- Zhang X, Yashiro M, Ohira M, et al. Synergic antiproliferative effect of DNA methyltransferase inhibitor in combination with anticancer drugs in gastric carcinoma. Cancer Sci. 2006;97:938–944. doi: 10.1111/j.1349-7006.2006.00253.x
- Lavoie H, Gagnon J, Therrien M. ERK signalling: a master regulator of cell behaviour, life and fate, nature reviews. Mol Cell Biol. 2020;21(10):607–632. doi: 10.1038/s41580-020-0255-7
- Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges, nature reviews. Drug Discovery. 2014;13:928–942. doi: 10.1038/nrd4281
- Lawrence MC, Jivan A, Shao C, et al. The roles of MAPKs in disease. Cell Res. 2008;18:436–442. doi: 10.1038/cr.2008.37
- Kamisawa T, Wood LD, Itoi T, et al. Pancreatic cancer. Lancet. 2016;388(10039):73–85. doi: 10.1016/S0140-6736(16)00141-0
- Nagarajan N, Bertrand D, Hillmer AM, et al. Whole-genome reconstruction and mutational signatures in gastric cancer. Genome Bio. 2012;13:R115. doi: 10.1186/gb-2012-13-12-r115
- Wong GS, Zhou J, Liu JB, et al. Targeting wild-type KRAS-amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat Med. 2018;24:968–977. doi: 10.1038/s41591-018-0022-x
- Liu H, Zhang Z, Han Y, et al. The FENDRR/FOXC2 axis contributes to multidrug resistance in gastric cancer and correlates with poor prognosis. Front Oncol. 2021;11:634579. doi: 10.3389/fonc.2021.634579
- Lu R, Wang X, Chen ZF, et al. Inhibition of the extracellular signal-regulated kinase/mitogen-activated protein kinase pathway decreases DNA methylation in colon cancer cells. J Biol Chem. 2007;282(16):12249–12259. doi: 10.1074/jbc.M608525200
- Issa JP. Optimizing therapy with methylation inhibitors in myelodysplastic syndromes: dose, duration, and patient selection. Nat Clin Pract Oncology. 2005;2(1):S24–29. doi: 10.1038/ncponc0355
- Thomas X. DNA methyltransferase inhibitors in acute myeloid leukemia: discovery, design and first therapeutic experiences. Expert Opin Drug Discov. 2012;7(11):1039–1051. doi: 10.1517/17460441.2012.722618
- Connolly RM, Li H, Jankowitz RC, et al. Combination epigenetic therapy in advanced breast cancer with 5-azacitidine and Entinostat: A Phase II National cancer Institute/Stand up to cancer study. Clin Cancer Res: An Official J Am Ass Cancer Res. 2017;23(11):2691–2701. doi: 10.1158/1078-0432.CCR-16-1729
- Overman MJ, Morris V, Moinova H, et al. Phase I/II study of azacitidine and capecitabine/oxaliplatin (CAPOX) in refractory CIMP-high metastatic colorectal cancer: evaluation of circulating methylated vimentin. Oncotarget. 2016;7:67495–67506. doi: 10.18632/oncotarget.11317
- Lee V, Wang J, Zahurak M, et al. A Phase I trial of a guadecitabine (SGI-110) and irinotecan in metastatic colorectal cancer patients previously exposed to irinotecan. Clin Cancer Res. 2018;24:6160–6167. doi: 10.1158/1078-0432.CCR-18-0421
- Juergens RA, Wrangle J, Vendetti FP, et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discovery. 2011;1:598–607. doi: 10.1158/2159-8290.CD-11-0214
- Tsai HC, Li H, Van Neste L, et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell. 2012;21(3):430–446. doi: 10.1016/j.ccr.2011.12.029
- Stewart ML, Tamayo P, Wilson AJ, et al. KRAS genomic status predicts the sensitivity of ovarian cancer cells to decitabine. Cancer Res. 2015;75(14):2897–2906. doi: 10.1158/0008-5472.CAN-14-2860
- Beyrouthy MJ, Garner KM, Hever MP, et al. High DNA methyltransferase 3B expression mediates 5-aza-deoxycytidine hypersensitivity in testicular germ cell tumors. Cancer Res. 2009;69(24):9360–9366. doi: 10.1158/0008-5472.CAN-09-1490
- Yu J, Qin B, Moyer AM, et al. DNA methyltransferase expression in triple-negative breast cancer predicts sensitivity to decitabine. J Clin Investig. 2018;128(6):2376–2388. doi: 10.1172/JCI97924
- Stresemann C, Brueckner B, Musch T, et al. Functional diversity of DNA methyltransferase inhibitors in human cancer cell lines. Cancer Res. 2006;66(5):2794–2800. doi: 10.1158/0008-5472.CAN-05-2821
- Stresemann C, Lyko F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer. 2008;123:8–13. doi: 10.1002/ijc.23607
- Serra RW, Fang M, Park SM, et al. A KRAS-directed transcriptional silencing pathway that mediates the CpG island methylator phenotype. Elife. 2014;3:e02313. doi: 10.7554/eLife.02313
- Wajapeyee N, Malonia SK, Palakurthy RK, et al. Oncogenic RAS directs silencing of tumor suppressor genes through ordered recruitment of transcriptional repressors. Genes Dev. 2013;27(20):2221–2226. doi: 10.1101/gad.227413.113
- Huang J, Stewart A, Maity B, et al. RGS6 suppresses Ras-induced cellular transformation by facilitating Tip60-mediated Dnmt1 degradation and promoting apoptosis. Oncogene. 2014;33(27):3604–3611. doi: 10.1038/onc.2013.324
- Gazin C, Wajapeyee N, Gobeil S, et al. An elaborate pathway required for Ras-mediated epigenetic silencing. Nature. 2007;449(7165):1073–1077. doi: 10.1038/nature06251
- Calvisi DF, Ladu S, Conner EA, et al. Inactivation of Ras GTPase-activating proteins promotes unrestrained activity of wild-type Ras in human liver cancer. J Hepatol. 2011;54(2):311–319. doi: 10.1016/j.jhep.2010.06.036
- Ordway JM, Williams K, Curran T. Transcription repression in oncogenic transformation: common targets of epigenetic repression in cells transformed by Fos, Ras or Dnmt1. Oncogene. 2004;23(21):3737–3748. doi: 10.1038/sj.onc.1207483
- Ma TM, Sun LP, Dong NN, et al. Protein expression trends of DNMT1 in gastrointestinal diseases: from benign to precancerous lesions to cancer. World J Gastrointest Oncol. 2019;11:1141–1150. doi: 10.4251/wjgo.v11.i12.1141
- Niwa T, Tsukamoto T, Toyoda T, et al. Inflammatory processes triggered by Helicobacter pylori infection cause aberrant DNA methylation in gastric epithelial cells. Cancer Res. 2010;70:1430–1440. doi: 10.1158/0008-5472.CAN-09-2755
- Niwa T, Toyoda T, Tsukamoto T, et al. Prevention of Helicobacter pylori-induced gastric cancers in gerbils by a DNA demethylating agent. Cancer Prev Res. 2013;6:263–270. doi: 10.1158/1940-6207.CAPR-12-0369
- Liao X, Siu MK, Chan KY, et al. Hypermethylation of RAS effector related genes and DNA methyltransferase 1 expression in endometrial carcinogenesis. Int J Cancer. 2008;123:296–302. doi: 10.1002/ijc.23494
- Calvisi DF, Ladu S, Gorden A, et al. Mechanistic and prognostic significance of aberrant methylation in the molecular pathogenesis of human hepatocellular carcinoma. J Clin Investig. 2007;117(9):2713–2722. doi: 10.1172/JCI31457
- Jin H, Wang X, Ying J, et al. Epigenetic silencing of a Ca 2+ -regulated Ras GTPase-activating protein RASAL defines a new mechanism of Ras activation in human cancers. Proc Natl Acad Sci, USA. 2007;104:12353–12358. doi: 10.1073/pnas.0700153104