
ABSTRACT
Background: Elucidating epigenetic mechanisms could provide new biomarkers for disease diagnosis and prognosis. Technological advances allow genome-wide profiling of 5-hydroxymethylcytosines (5hmC) in liquid biopsies. 5hmC-Seal followed by NGS is a highly sensitive technique for 5hmC biomarker discovery in cfDNA. Currently, 5hmC Seal is optimized for EDTA blood collection. We asked whether heparin was compatible with 5hmC Seal as many clinical and biobanked samples are stored in heparin.
Methods: We obtained 60 samples in EDTA matched to 60 samples in heparin from the Prostate, Lung, Colorectal, and Ovarian (PLCO) Cancer Screening Trial. Samples were comprised of 30 controls and 30 individuals who were later diagnosed with colon cancer. We profiled genome-wide 5hmC in cfDNA using 5hmC-Seal assay followed by NGS. The 5hmC profiling data from samples collected in EDTA were systematically compared to those in heparin across various genomic features.
Results: cfDNA isolation and library construction appeared comparable in heparin vs. EDTA. Typical genomic distribution patterns of 5hmC, including gene bodies and enhancer markers, were comparable in heparin vs. EDTA. 5hmC analysis of cases and controls yielded highly correlated differential features suggesting that both anticoagulants were compatible with 5hmC Seal assay.
Conclusions: While not currently recommended for the 5hmC-Seal protocol, blood samples stored in heparin were successfully used to generate analysable and biologically relevant genome-wide 5hmC profiling. Our findings are the first to support opportunities to expand the biospecimen resource to heparin samples for 5hmC Seal and perhaps other PCR-based technologies in epigenetic research.
Introduction
Epigenetic modifications play critical roles in gene regulation and disease development. Besides the more widely investigated DNA methylation (i.e., 5-methylcytosines or 5mC), DNA hydroxymethylation (i.e., 5-hydroxymethylcytosines or 5hmC) at CpG dinucleotides has been demonstrated to be relevant to gene regulation in various normal and pathological processes [Citation1,Citation2]. Technological advances have allowed the interrogation of genome-wide 5hmC in clinically relevant biospecimens, such as circulating cell-free DNA (cfDNA) isolated from peripheral blood for clinical applications [Citation2,Citation3]. Of note, the 5hmC-Seal [Citation4,Citation5], a highly sensitive chemical labelling technique when coupled with NGS, is a powerful tool for biomarker discovery in circulating cfDNA. This assay requires, for example, only a few nanograms of cfDNA from <5 mL of plasma. Our laboratory and others have applied the 5hmC-Seal technique in cfDNA to explore diagnostic and prognostic biomarkers for a wide range of human diseases, including cancers [Citation6–12], cardiovascular diseases [Citation13], and diabetic complications [Citation14–17], as well as 5hmC underlying cancer subtypes and population differences [Citation18,Citation19].
The current protocol recommends the use of EDTA-coated blood collection tubes for the assay as heparin can interfere with polymerase chain reaction (PCR) [Citation20], a step in the 5hmC-Seal library construction required to amplify chemically labelled 5hmC-containing cfDNA fragments. Characterizing 5hmC-Seal profiling data generated from blood samples stored in heparin-coated tubes could provide valuable information about whether this powerful technique could utilize heparinized biospecimens that are widely available in biobanks and other clinical resources.
In the current study, we applied the 5hmC-Seal technique to cfDNA isolated from 120 plasma samples stored in EDTA- or heparin-coated tubes obtained from the Prostate, Lung, Colorectal and Ovarian (PLCO) Cancer Screening Trial [Citation21], a tremendous resource of clinical biospecimens for biomarker discovery. The genome-wide 5hmC profiles generated from these samples were systematically compared across various genomic features to assess the performance of 5hmC-Seal profiling in heparin vs. EDTA stored blood samples. Findings from this comparison study provided valuable information about cfDNA preparation, library construction, genomic coverage, and genome-wide 5hmC distribution for blood samples stored heparin vs. EDTA. These results serve to expand the sample pool applicable for assay by this powerful technique in clinical epigenetic research.
Methods
PLCO plasma samples
We designed this study to include PLCO trial samples collected from study subjects between 1998 and 2004 under our collaborative contract with the National Cancer Institute (U01CA217078). Because race is a confounder in genome-wide 5hmC data analysis, we requested samples from PLCO study participants of non-Hispanic European ancestry. We requested n = 120 paired (EDTA and heparin) plasma samples collected from n = 60 study participants (). Among the 60 individuals, 30 of them were diagnosed later with colorectal cancer within 1 y of the 3rd sampling (i.e., T3 time point) of the PLCO Trial [Citation21], and 30 individuals were age-, gender-, and race/ethnicity- matched controls from the same T3 sampling time point (Supplementary Table S1). The controls were study participants who did not develop overt cancer followed for 15 y after the T3 blood collection time point. All the demographic and clinical variables were de-identified for these samples.
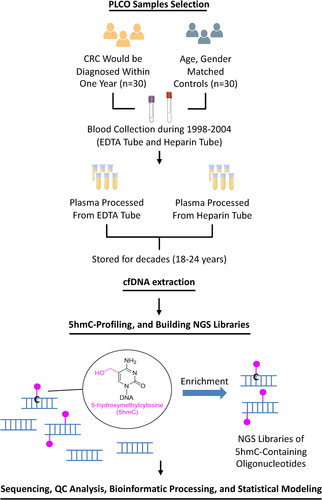
Figure 1. Study design and workflow. the workflow includes PLCO sample selection, cfDNA extraction, 5hmC-Seal profiling, bioinformatic processing, and statistical modelling.
Isolation of cfDNA from plasma samples
cfDNA was extracted from 0.3 ml plasma per PLCO sample using QIAamp Circulating Nucleic Acid Kit (Qiagen, Germantown, Maryland) with elution into nuclease-free water following the manufacturer’s protocol. The cfDNA concentrations were quantitated using the Qubit dsDNA High Sensitivity Assay™ (Thermo Fisher Scientific, Waltham, MA).
5hmC-Seal library construction and sequencing
Following our established protocol, 8 ng cfDNA was used for 5hmC-Seal library construction [Citation7]. Briefly, after end-repair and A-tailing using KAPA Hyper Prep Kit (Roche, Indianapolis, IN) and ligation of the KAPA Unique Dual-Indexed Adapter (Roche), the product was purified using DCC-5 Clean and Concentrator (Zymo Research, Irvine, CA). 5hmC modifications were labelled with UDP-azide-glucose (Active Motif, Carlsbad, California) and T4 β-glucosyltransferase (βGT) enzyme (Thermo Fisher Scientific). The purified product (DCC-5 Clean and Concentrate, Zymo) was reacted with DBCO-PEG4-biotin (Active Motif). Biotinylated DNA was subsequently pulled down and enriched by binding to magnetic streptavidin beads (Dynabeads M-270, Invitrogen, Waltham, MA). The NGS libraries were then constructed using KAPA Hyper Prep Kit (Roche) with on-bead PCR amplification and subsequently purified with AMPure XP Beads (Beckman Coulter, Indianapolis, IN). All libraries were QC checked by fragment analyser, normalized, and sequenced using paired-end mode (PE50) at The University of Chicago Functional Genomics Facility using the NovaSeq6000 Platform (Illumina, San Diego, CA).
Bioinformatic processing
Bioinformatics analyses were carried out as described in our previous publications [Citation7–9]. Briefly, adapter sequences were removed from raw sequencing reads using Trim_Galore [Citation22]. Low-quality bases at the 5’ (phred score < 5) and 3’ (5 bp-sliding window phred score < 15) were trimmed to a minimum length of 30 bp. Sequencing reads were aligned to the human genome reference (hg19) using Bowtie2 with end-to-end alignment mode [Citation23]. Read pairs were concordantly aligned with fragment length ≤500 bp and an average ≤1 ambiguous base and up to four mismatched bases per 100 bp length. Alignments with Mapping Quality Score ≥10 were counted for overlap with GENCODE [Citation24] gene bodies using featureCounts [Citation25] without strand information. 5hmC profiles were summarized for enhancer markers H3K4me1 and H3K27ac derived from GM12878 as provided by the Encyclopedia of DNA Elements (ENCODE) Project [Citation26]. All 120 raw sequencing read counts were normalized using DESeq2 [Citation27], which performs an internal normalization that corrects for library size and were included in downstream analyses. The raw and processed 5hmC-Seal sequencing data are available to investigators with appropriate requests.
Differential analysis and simulation
To evaluate whether samples stored in heparin vs. EDTA would generate correlated results, we compared genomic 5hmC findings between 30 participants who were later diagnosed with CRC (cases), and 30 age-, race-, and gender-matched controls who did not develop overt cancers followed for up to 15 y. Briefly, the normalized 5hmC levels (i.e., read counts summarized for gene bodies) were compared between cases and controls using DESeq2 [Citation27] in the EDTA subset and separately in the heparin subset. We performed a simulation analysis to estimate the significance of shared top differential gene bodies between cases and controls as detected in samples stored in heparin vs. EDTA. We then generated a null distribution. Specifically, for each N number of observed top differential genes (e.g., the N top differential genes obtained from EDTA and heparin samples), the same number of N genes is randomly selected from the whole genome background, and the null distribution was constructed by repeating this procedure 10,000 times.
Results
cfDNA quantification and construction of 5hmC-Seal libraries
After cfDNA was isolated from plasma samples, we quantified cfDNA and observed a trend towards higher concentrations of total cfDNA from the heparin plasma (). This trend was consistent between cases and controls (), though the trend is more significant for cases than controls. We also analysed the amount and percentage of libraries with a 200–1000 bp range generated from cfDNA fragments. The pattern of the amount and the size distribution of libraries were comparable between samples collected in EDTA- vs. heparin-coated tubes (), which was also consistently observed in cases and controls (). The resulting 5hmC-Seal libraries showed similar electrophoresis patterns in heparin vs. EDTA. shows that we could construct 5hmC-Seal libraries from cfDNA isolated from plasma samples stored in ETDA- and heparin-coated tubes. Notably, library sizes also appeared to be consistent comparing samples stored in heparin vs. EDTA (). Two random pairs of samples stored in EDTA vs. heparin show that the size and distributions of the 5hmC-Seal libraries were comparable in the two collection tubes ().
Figure 2. Quantification of cfDNA and NGS libraries.
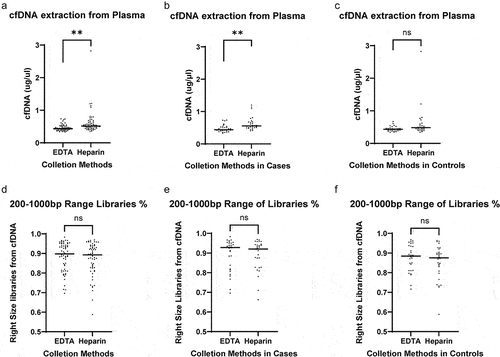
Figure 3. Distribution of the 5hmC-Seal libraries.
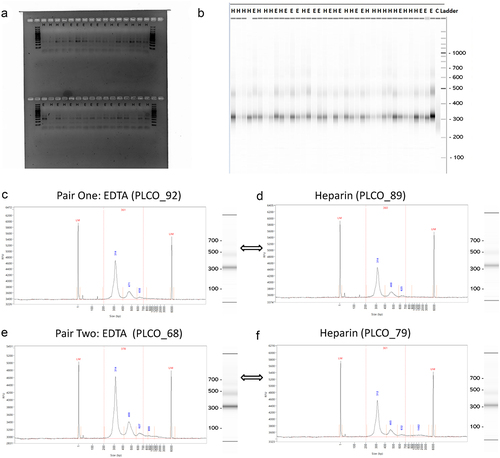
Overview of the 5hmC-Seal sequencing data
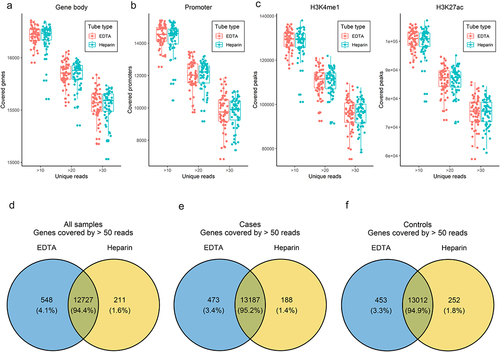
Overall, the duplication rates between samples stored in EDTA vs. heparin were comparable (Supplementary Figure S1), likely reflecting similar levels of genomic complexity (i.e., unique cfDNA fragments) under the different storage conditions. Of note, PCA plots showed no systematic bias observed in the genome-wide 5hmC profiles as summarized by gene bodies across all cfDNA samples by anticoagulant () or stratified by diagnosis (case, control), age group, or gender (). We further compared the genomic coverage between samples stored in different anticoagulants. Consistent across a series of cut-offs for unique reads (i.e., >10, >20, and >30), in terms of gene bodies, the overall coverage was comparable between samples stored in EDTA- vs. heparin-coated tubes (). Similar observations were made for promoters and histone modifications marking enhancers (H3K4me1 and H3K27ac) (). Furthermore, for those gene bodies with >50 unique reads, the majority of the covered gene bodies were shared between samples stored in different anticoagulants (). For example, 13,187 gene bodies, 95.2% in total, were well covered in cases and 13,012 in controls, 94.9% in total, comparing samples collected in heparin vs. EDTA ().
Figure 4. Genome-wide 5hmC profiles between samples stored in different anticoagulants.
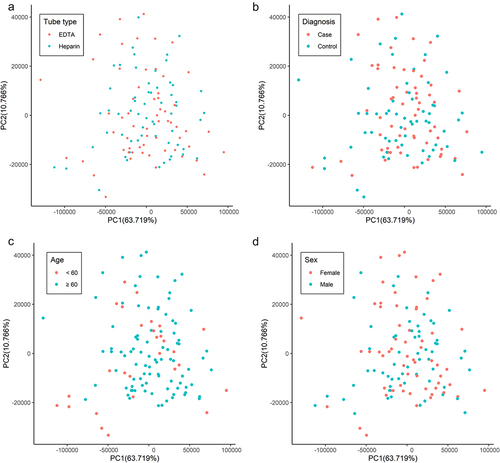
Figure 5. Comparison of genomic coverage by unique reads.
Comparison of the distribution of genome-wide 5hmC-Seal profiles
Typical 5hmC genomic distributions were observed comparing heparin vs. EDTA. Specifically, gene bodies exhibited higher 5hmC modification levels compared to flanking regions, lower modification levels at promoters, and enrichment at enhancers, as identified by histone modification marks (), which were also consistent in latent colorectal cases and controls separately (). For the primary genomic feature used in previous 5hmC-Seal studies, i.e., gene bodies, we showed that samples matched to the same individual but stored in different anticoagulants had an overall higher correlation than sample pairs from non-matched individuals (Pearson’s Correlation: 0.988 ± 0.00965 sd. vs. 0.981 ± 0.0119 sd.) ().
Figure 6. Distribution of genome-wide 5hmC and correlation.
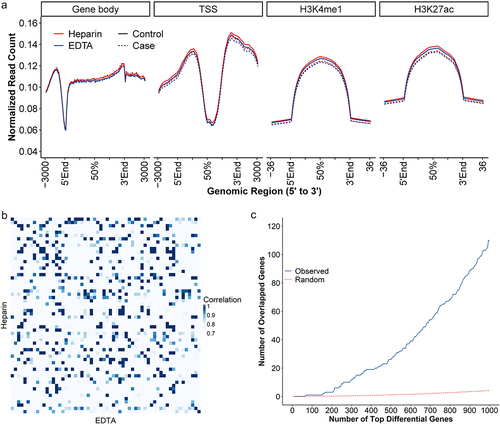
Using a simulation analysis, we further showed that the differential results (cases vs. controls) were significantly shared between samples stored in different anticoagulants (). For example, the top 1000 differential genes showed ~10-fold enrichment in overlapping results between samples stored in EDTA vs. heparin relative to the null distribution.
Discussion
Our primary objective was to compare the results of 5hmC-Seal analysis for patient-matched samples stored in heparin vs. EDTA from the PLCO trial. We realized that the PLCO samples were decades old and at risk for deterioration in DNA quality because of the long storage time. A direct comparison between matched samples stored in different anticoagulants collected at the same time point, however, allowed us to focus on the potential impact of heparin vs. EDTA on the quality of 5hmC-Seal data.
Before comparing genome-wide 5hmC profiles, we showed that although the extracted cfDNA concentrations from heparinized samples were higher than matched samples stored in EDTA-coated tubes, there were no differences in concentrations within the expected molecular weight range of 5hmC-Seal libraries (200–1000 nt) built from cfDNA fragments. Notably, the 5hmC-Seal library construction yielded similar electrophoretic patterns (e.g., size and distribution) for cfDNA from EDTA- vs. heparin-coated tubes suggesting comparable library construction efficiency. This alleviates concerns about heparin inhibition of DNA polymerases and supports the feasibility of expanding the 5hmC-Seal protocol to include heparin-stored samples.
We then systematically compared genome-wide 5hmC profiles between cfDNA stored in EDTA vs. heparin. Firstly, the genomic distributions of the 5hmC profiles showed typical patterns (i.e., enriched in gene bodies and regions with activation transcription enhancer marks) as reported in our previous studies [Citation7,Citation9], regardless of diagnosis (cases vs. controls), indicating successful profiling of 5hmC in these samples. As noted, the PLCO samples were collected decades ago (~20 y), and biodegradation (e.g., DNA degradation) could occur. Systematic analyses of gene bodies, i.e., the primary genomic feature used in previous studies, with a series of cut-offs for unique reads (10, 20, 30, and 50) showed that there were still ~12–13,000 gene bodies out of ~19,000 genes covered by at least 50 unique reads, supporting the feasibility of performing statistical analyses and modelling using these data. The fact that both cases and controls showed comparable genome-wide 5hmC patterns when analysing samples stored in EDTA vs. heparin further supported the robustness of the 5hmC-Seal profiling methodology for epigenetic analysis of these samples.
Since a primary utility of the 5hmC-Seal technique in cfDNA is biomarker discovery, we further separately compared the differential results obtained between cases and controls in samples stored in EDTA vs. heparin. Although the primary goal of the current study was not biomarker discovery, we showed that the differential genes for samples stored in EDTA vs. heparin showed significant overlap relative to the null distribution via simulation analysis. This further supports our conjecture that we can obtain meaningful biological information from these samples whether stored in heparin or EDTA.
There are some limitations of the current study. Firstly, as we emphasized, the depth of our analysis has certain limitations due to the extensive duration of sample storage, with the PLCO samples having been collected years ago, raising concerns about potential DNA degradation over time. Distinct variations may be present when comparing aged samples with their freshly collected counterparts, or samples subject to shorter freezing durations, or those exposed to less freeze–thaw cycles. Hence, a comparison in freshly collected samples stored in EDTA vs. heparin would likely provide a more sensitive view of potential difference between the two storage conditions and confirm the current findings. Secondly, the cases from the PLCO Trial are pre-diagnostic. Therefore, the comparison of 5hmC differences between the PLCO cases and controls was not intended to represent a comparison between colorectal cancer patients and healthy controls. None of the cases had overt colon cancer. Because the primary aim of this study was a technical comparison between EDTA vs. heparin, we reasoned that the distinction between case and control would not affect the conclusion we drew from the current study. Our results support this conclusion.
Taken together, our systematic comparison between cfDNA derived from PLCO samples stored in EDTA vs. heparin suggests that expanding the 5hmC-Seal application from EDTA-stored samples to heparin-stored samples is feasible, based upon supportive evidence including comparable cfDNA concentration for target fragments, library size, genomic coverage, 5hmC genomic distribution, and differential results between cases and controls. Our findings demonstrate the potential for broadening the 5hmC-Seal assay for biomarker discovery and clinical epigenetic research to wider storage conditions and potentially increase the value of the PLCO biorepository and many other clinical and biobanked samples.
Supplemental Material
Download Zip (384.7 KB)Acknowledgments
We thank the National Cancer Institute (NCI) for access to NCI’s data collected by the Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial who was funded in whole or in part with federal funds from the NCI, US National Institutes of Health (NIH).
Disclosure statement
C.H. is an investigator at the Howard Hughes Medical Institute. C.H.is ascientific founder and ascientific advisory board member of Accent Therapeutics, Inc., and Inferna Green, Inc. All other authors declare they have no competing interests.
Data availability statement
The raw and processed 5hmC-Seal data have been deposited into the NCBI Gene Expression Omnibus: Accession No. GSE230027.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15592294.2023.2271692.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Branco MR, Ficz G, Reik W. Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nat Rev Genet. 2011;13(1):7–11. doi: 10.1038/nrg3080
- Zeng C, Stroup EK, Zhang Z, et al. Towards precision medicine: advances in 5-hydroxymethylcytosine cancer biomarker discovery in liquid biopsy. Cancer Commun (Lond). 2019;39(1):12. doi: 10.1186/s40880-019-0356-x
- Liu M, Zhang Z, Zhang W, et al. Advances in biomarker discovery using circulating cell-free DNA for early detection of hepatocellular carcinoma. WIREs Mechanisms of Disease. 2023;15(3):e1598. doi: 10.1002/wsbm.1598
- Song CX, Szulwach KE, Fu Y, et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nature Biotechnol. 2011;29(1):68–72. doi: 10.1038/nbt.1732
- Han D, Lu X, Shih AH, et al. A highly sensitive and robust method for genome-wide 5hmC profiling of rare cell populations. Molecular Cell. 2016;63(4):711–719. doi: 10.1016/j.molcel.2016.06.028
- Gao P, Lin S, Cai M, et al. 5-hydroxymethylcytosine profiling from genomic and cell-free DNA for colorectal cancers patients. J Cell Mol Med. 2019;23(5):3530–3537. doi: 10.1111/jcmm.14252
- Li W, Zhang X, Lu X, et al. 5-hydroxymethylcytosine signatures in circulating cell-free DNA as diagnostic biomarkers for human cancers. Cell Res. 2017;27(10):1243–1257. doi: 10.1038/cr.2017.121
- Cai J, Zeng C, Hua W, et al. An integrative analysis of genome-wide 5-hydroxymethylcytosines in circulating cell-free DNA detects noninvasive diagnostic markers for gliomas. Neurooncol Adv. 2021;3(1):vdab049. doi: 10.1093/noajnl/vdab049
- Cai J, Chen L, Zhang Z, et al. Genome-wide mapping of 5-hydroxymethylcytosines in circulating cell-free DNA as a non-invasive approach for early detection of hepatocellular carcinoma. Gut. 2019;68(12):2195–2205. doi: 10.1136/gutjnl-2019-318882
- Song CX, Yin S, Ma L, et al. 5-hydroxymethylcytosine signatures in cell-free DNA provide information about tumor types and stages. Cell Res. 2017;27(10):1231–1242. doi: 10.1038/cr.2017.106
- Chiu BC, Zhang Z, You Q, et al. Prognostic implications of 5-hydroxymethylcytosines from circulating cell-free DNA in diffuse large B-cell lymphoma. Blood Adv. 2019;3(19):2790–2799. doi: 10.1182/bloodadvances.2019000175
- Applebaum MA, Barr EK, Karpus J, et al. 5-hydroxymethylcytosine profiles in circulating cell-free DNA associate with disease Burden in children with neuroblastoma. Clin Cancer Res. 2020;26(6):1309–1317. doi: 10.1158/1078-0432.CCR-19-2829
- Dong C, Chen J, Zheng J, et al. 5-hydroxymethylcytosine signatures in circulating cell-free DNA as diagnostic and predictive biomarkers for coronary artery disease. Clin Epigenetics. 2020;12(1):17. doi: 10.1186/s13148-020-0810-2
- Zeng C, Yang Y, Zhang Z, et al. 304-OR: 5-hydroxymethylcytosines in circulating cell-free DNA Reveal Diabetic nephropathy. Diabetes. 2020;69(Supplement_1):304–OR. doi: 10.2337/db20-304-OR
- Yang Y, Zeng C, Lu X, et al. 5-hydroxymethylcytosines in circulating cell-free DNA reveal vascular complications of type 2 diabetes. Clin Chem. 2019;65(11):1414–1425. doi: 10.1373/clinchem.2019.305508
- Han L, Chen C, Lu X, et al. Alterations of 5-hydroxymethylcytosines in circulating cell-free DNA reflect retinopathy in type 2 diabetes. Genomics. 2020;113(1 Pt 1):79–87. doi: 10.1016/j.ygeno.2020.11.014
- Zhang Z, Beadell A, Capuano A, et al. PB2428: genome-wide mapping implicates 5-hydroxymethylcytosines in diabetes and Alzheimer's disease. In: The ASHG Annual Meeting; 2022; Los Angeles, CA;
- Chiu BC, Zhang Z, Derman BA, et al. Genome-wide profiling of 5-hydroxymethylcytosines in circulating cell-free DNA reveals population-specific pathways in the development of multiple myeloma. J Hematol Oncol. 2022;15(1):106. doi: 10.1186/s13045-022-01327-y
- Chiu BC, Chen C, You Q, et al. Alterations of 5-hydroxymethylation in circulating cell-free DNA reflect molecular distinctions of subtypes of non-Hodgkin lymphoma. NPJ Genom Med. 2021;6(1):11. doi: 10.1038/s41525-021-00179-8
- Yokota M, Tatsumi N, Nathalang O, et al. Effects of heparin on polymerase chain reaction for blood white cells. J Clin Lab Analysis. 1999;13(3):133–140. doi: 10.1002/(SICI)1098-2825(1999)13:3<133:AID-JCLA8>3.0.CO;2-0
- Zhu CS, Pinsky PF, Kramer BS, et al. The prostate, lung, colorectal, and ovarian cancer screening trial and its associated research resource. JNCI. 2013;105(22):1684–1693. doi: 10.1093/jnci/djt281
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170
- Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923
- Harrow J, Frankish A, Gonzalez JM, et al. GENCODE: the reference human genome annotation for the ENCODE project. Genome Res. 2012;22(9):1760–1774. doi: 10.1101/gr.135350.111
- Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–930. doi: 10.1093/bioinformatics/btt656
- An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi: 10.1038/nature11247
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8