
ABSTRACT
Smoking is a potent cause of asthma exacerbations, chronic obstructive pulmonary disease (COPD) and many other health defects, and changes in DNA methylation (DNAm) have been identified as a potential link between smoking and these health outcomes. However, most studies of smoking and DNAm have been done using blood and other easily accessible tissues in humans, while evidence from more directly affected tissues such as the lungs is lacking. Here, we identified DNAm patterns in the lungs that are altered by smoking. We used an established mouse model to measure the effects of chronic smoke exposure first on lung phenotype immediately after smoking and then after a period of smoking cessation. Next, we determined whether our mouse model recapitulates previous DNAm patterns observed in smoking humans, specifically measuring DNAm at a candidate gene responsive to cigarette smoke, Cyp1a1. Finally, we carried out epigenome-wide DNAm analyses using the newly released Illumina mouse methylation microarrays. Our results recapitulate some of the phenotypes and DNAm patterns observed in human studies but reveal 32 differentially methylated genes specific to the lungs which have not been previously associated with smoking. The affected genes are associated with nicotine dependency, tumorigenesis and metastasis, immune cell dysfunction, lung function decline, and COPD. This research emphasizes the need to study CS-mediated DNAm signatures in directly affected tissues like the lungs, to fully understand mechanisms underlying CS-mediated health outcomes.
Introduction
Smoking is one of the leading causes of preventable mortality worldwide, and poses a great burden on the healthcare system, as it has been linked to the development and poor clinical control of diseases like asthma, chronic obstructive pulmonary disease (COPD), bronchitis, obesity, and cancers [Citation1–5]. Epigenetic mechanisms, particularly DNA methylation (DNAm), have been suggested as potential links between environmental exposures such as smoking and disease outcomes that might manifest later in life.
There is abundant evidence that smoking alters DNAm patterns in human peripheral blood [Citation6–9] and leucocyte subtypes [Citation7], lymphoblasts and pulmonary macrophages [Citation10], and epithelial oral cells [Citation11]. In fact, there are reports that methylation levels of certain CpGs are effective markers for predicting smoking status [Citation12] and lung cancer incidence [Citation13]. However, most of these past studies associating smoking with DNAm alterations were done in accessible tissues, such as blood or epithelia, where large-scale sample collections are possible. Data from large human studies using accessible tissues is extremely helpful in identifying potential biomarkers of exposure or outcome, but evidence from more proximal tissues such as lungs which could develop our mechanistic understanding is severely lacking [Citation14]. DNAm is cell type and tissue-specific, and so it is not clear whether findings from peripheral tissues like blood can be applied to the lungs [Citation15–18]. One recent study measured epigenome-wide DNAm in current smokers and reported that only a third of the significantly altered CpGs which they originally identified in whole blood were also altered in the lungs of current compared to never-smokers [Citation14]. Another study compared smoking-induced DNAm changes in blood versus buccal cells and reported that DNAm sites from buccal cells outperformed those identified in blood cells in discrimination of 14 of 15 epithelial cancer types [Citation19]. The latter study showed that tissues directly exposed to cigarette smoke (CS) such as lungs, buccal cells and saliva may demonstrate more similar effects of smoking on DNAm, but it is clear that for truly mechanistic understanding, it is essential to examine the main tissues of interest. It can be difficult to access these tissues in humans, so model organisms are often employed.
Studies using animal models to measure the effects of smoking on DNAm in the lungs are also limited [Citation14,Citation20,Citation21]. One study measured the effect of 4 weeks of smoking on lung DNAm using capture-based bisulphite sequencing and found altered DNAm patterns for numerous genes, most involved in inflammation and in inflammatory injury in COPD [Citation21]. Another measured epigenome-wide DNAm in the blood of current and never-smokers and then measured the corresponding DNAm at the significant CpGs in the lungs via pyrosequencing [Citation14]. Their results revealed that only one-third significant CpGs in the blood also showed changes in DNAm in the lungs, demonstrating the importance of assessing DNAm in the specific tissue of interest [Citation14]. A third study measured the effects of whole-body CS, tobacco carcinogens and lipopolysaccharides in mouse lungs using reduced representation bisulphite sequencing (RRBS) [Citation20]. They reported DNAm and hydroxymethylation differences across multiple differentially methylated regions [Citation20]. These studies are few and thus indicate that there is a clear need for more research on the effects of smoking on DNAm patterns in lung tissues.
DNAm microarray is a high-throughput method for epigenome-wide DNAm measurements at a lower cost and is less labour intensive than bisulphite sequencing. Sequentially increasing scales of the Infinium microarrays have been developed for human samples, but no such solution was available for use on mouse samples until recently, with the release of the Illumina Infinium mouse microarrays [Citation22]. In the current study, we used an animal model of chronic smoking to measure effects of smoking on lung function, as well as gene expression and DNAm of a candidate gene, both immediately after smoking and 15 weeks after smoking cessation. We also employed the new mouse methylation array to measure epigenome-wide DNAm immediately after smoking. Our model successfully recapitulated some of the lung phenotypes and DNAm signatures observed in humans. It also reveals many novel sites in the lungs which are differentially methylated by smoking, involved in nicotine dependence, altered immune response and development of malignancies, lung function decline and COPD, cardiovascular defects and neurodegenerative disorders. This study is an essential step in understanding mechanisms underlying CS-induced health alterations, and therefore would help in developing therapies to mitigate the effects of smoking.
Methods
Animals and smoking model
This experiment was approved by the Animal Research Ethics and Compliance Committee of the University of Manitoba (protocol number 19–021 (AC11461)). Adult BALB/c mice (Charles River Laboratories, Massachusetts, United States) were given standard laboratory chow and clean water ad libitum and housed, four mice of a single sex per cage (except where noted), in individually ventilated cages in a 12-hour light/dark cycle throughout the duration of this experiment.
The full details of the smoke exposure paradigm have been published previously [Citation23]. Briefly, 8-week-old adult female BALB/c mice were separated into two groups: 16 control mice and 16 smoke-exposed mice. Treatments were for a total period of 9 weeks, beginning 3 weeks prior to mating (at which point mice were singly housed), continuing throughout pregnancy and lasting until 3 weeks after the birth of offspring. Control mice were moved to a clean cage and exposed to room air, while CS mice were exposed to whole-body 1R6F research cigarettes (University of Kentucky, Lexington, KY), twice daily using the SCIREQ InExpose smoking robot (SCIREQ, Montreal, QC, Canada). The results reported in this study are a part of a larger experiment to investigate the effects of maternal smoking on offspring [Citation23]. Therefore, the ‘dams’ referred to from this point forward were all pregnant in line with that experiment. Dams were weighed weekly and 48 to 72 hours after the last smoke exposure (referred to hereafter as ‘immediately after smoking’), a subset of dams underwent lung function testing followed by tissue collection. The remaining dams underwent lung function testing and tissue collection 15 to 16 weeks after smoking cessation (referred to hereafter as ‘15 weeks after smoking cessation’), resulting in data from 2 timepoints: immediately after 9 weeks of smoking and 15 weeks after smoking cessation (Sample and group information can be found in Supplementary Table S1). All supplemental information is available at: https://doi.org/10.6084/m9.figshare.24546013.v3.
Measurement of maternal cotinine
To verify the delivery of CS, the cotinine levels were measured in plasma immediately after smoking, and again 15 weeks later in a subset of dams. Cotinine was measured using a commercially available ELISA kit (CalBiotech, Spring Valley, CA), according to the manufacturer’s instructions, and the concentration of cotinine in each sample was measured at 450 nm.
Lung function measurement and Bronchoalveolar lavage fluid collection
Mice were anaesthetised with sodium pentobarbital and lung function measured using the SCREQ Flexivent small animal ventilator (SCIREQ Inc., Montreal, QC, Canada) as described previously [Citation24,Citation25]. Four lung function metrics: Total airway resistance (Rrs), Newtonian resistance (Rn), tissue resistance (G) and elastance (H) were assessed, first at baseline after injection of nebulized saline into the lungs, and then after introduction of increasing concentrations of nebulized methacholine (3 to 50 mg/mL). Methacholine serves as a cholinergic agonist which acts on muscarinic receptors in airway smooth muscles, thereby inducing bronchoconstriction and decreasing lung function [Citation26,Citation27]. As a result, methacholine challenge tests are considered the gold standard for measuring airway hyperresponsiveness in mouse models [Citation26,Citation27].
Once lung function measurements were complete, mouse lungs were washed twice with 1 mL of phosphate buffer saline (PBS) per wash, introduced via tracheal cannulation. The bronchoalveolar lavage fluid (BALF) obtained was centrifuged at 4°C at 1200 rpm for 10 min, and the supernatants were stored at −20°C to be used for future analysis. Cell pellets were resuspended in 1 mL of PBS for total cell counts using a haemocytometer. Differential cell counts of macrophages, eosinophils and lymphocytes were performed by first pipetting 100 uL of PBS-resuspended cell pellets onto glass slides using cytospin columns, staining with a modified Wright-Giemsa stain (Hema 3 Stat Pack), and then counting cells using a Carl Zeiss Axio Observer ZI microscope.
Tissue collection, DNA/RNA isolation
We collected whole blood from dams immediately after 9 weeks of CS exposure and 15 weeks after smoking cessation. Blood obtained from the severed abdominal aorta was immediately pipetted into EDTA-coated tubes and centrifuged at 4000 rpm for 15 min to obtain plasma. Following separation, blood cell pellets and plasma were snap-frozen in dry ice and stored at −80°C. Next, left, middle, superior, inferior and postcaval lung lobes were collected into separate tubes and snap-frozen in dry ice.
To prepare for simultaneous DNA and RNA extraction using the Invitrogen DNA and RNA isolation kits, we homogenized whole left lungs in Qiagen RLT Plus buffer using the Qiagen Tissue Lyser II. We also extracted DNA from blood cell pellets using the Qiagen DNAeasy Blood and Tissue kit and quantified all extracted DNA and RNA using a NanoDrop spectrophotometer (NanoDrop Technologies, USA).
Selection of candidate genes to measure DNAm in mice
As our mouse model was novel, we needed to confirm that we could use it to recapitulate some of the DNAm signatures previously reported in humans exposed to CS. We decided to measure DNAm at candidate genes first on a small scale and then proceed to epigenome-wide DNAm measurements. Complete details of how we selected our two candidate genes have been published previously [Citation23]. Briefly, we selected two CpGs which were the most responsive to in utero CS in newborn umbilical cord blood samples as reported in a meta-analysis conducted across human cohorts, and had also been associated with exposure to CS in adulthood [Citation28]. The two chosen human CpGs were AHRR (cg05575921) and CYP1A1 (cg22549041). We then aligned these sequences against the GRCm38/mm10 Mus musculus genome assembly using muscle [Citation29] alignment on R studio version 3.6. This produced mouse CpGs which exactly aligned with or were closest in position to the human CpGs selected from the meta-analysis. The two mouse CpGs were at chr13:74260517 for Ahrr and chr9:57696231 for Cyp1a1 (GRCm38/mm10). It is essential to note that this region of Ahrr is not conserved between humans and mice, and while we selected the closest mouse CpG, it may not be comparable to the human position.
Gene expression measurement
200 ng of lung mRNA was converted to cDNA using Maxima cDNA synthesis Kit (Thermo Fisher Scientific, Inc.) following manufacturer’s protocol. We measured the relative expression of Ahrr and Cyp1a1 in dam lungs using quantitative real-time RT-PCR (qPCR) performed on the QuantStudio 3 Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA). Ahrr and Cyp1a1 expression was quantified using the ΔΔCq method [Citation30,Citation31] after normalizing against mean β-actin and Eif2a levels in the same sample. Samples were run in duplicates under cycling conditions that were recommended by the manufacturer, and the primer sequences used have been published previously [Citation23].
Measurement of DNAm at the candidate genes, Ahrr and Cyp1a1
We measured DNAm at the two chosen mouse candidate genes, Ahrr and Cyp1a1, via pyrosequencing, as previously described [Citation23]. Briefly, we performed bisulphite conversion (Zymo Research) on 500 ng of DNA isolated from the left lungs or blood to generate bisulphite-converted DNA (bcDNA), following the manufacturer’s protocol. Converted DNA was amplified using primers and conditions published previously [Citation23]. DNAm at candidate genes was measured in duplicates using the Qiagen Q48 pyrosequencer, alongside mouse control DNA of increasing methylation concentrations: 0%, 25%, 50%, 75%, and 100%.
Measurement of epigenome-wide DNAm in dam lung autosomes
We used the Illumina Infinium Mouse Methylation BeadChip (Illumina, San Diego, CA, USA) to measure DNAm across the whole mouse epigenome in accordance to the manufacturer’s protocol. Briefly, 750 ng of genomic DNA was bisulphite converted as described above. Next, bisulphite converted samples were randomized, amplified, fragmented, hybridized onto the array chip and scanned according to the standard protocol.
We exported IDAT files into R and performed preprocessing using the SeSAMe package [Citation32]. Preprocessing steps included removal of 18,920 of 296,070 probes mapping to the X, Y and mitochondrial chromosomes, calculation of detection p values per probe using pOOBAH [Citation32], background subtraction using noob [Citation33], dye bias correction using the dyeBiasCorrTypeINorm function in the SeSAMe package and filtering out probes with detection p > 0.05. Next, signal intensities of the remaining 208,606 probes were quantified as β-values and used in downstream analyses.
Measurement of epigenome-wide DNAm in dam lung X chromosomes
We measured the effect of smoking for 9 weeks on DNAm in Illumina mouse microarray probes mapping to the female X chromosomes separately. Similar to autosomal probes, preprocessing was conducted using the SeSAMe package in R [Citation32]. We performed the preprocessing steps highlighted above on 15,117 of 296,070 probes mapping to the female X chromosome, and were left with 11,529 probes used in downstream analyses.
Statistical analyses
Statistical analyses were performed in RStudio (versions 3.6.1 and 4.2). To analyse candidate gene pyrosequencing data, we averaged DNAm at each CpG across duplicates and measured between-group differences using student t-tests. We used one-way ANOVA to analyse lung function and differential cell count data, followed by student t-tests for multiple comparisons between groups at each methacholine dose. We considered p values <0.05 as significant.
To analyse epigenome-wide DNAm microarray data, we first performed chip, run, row and column batch correction on β values using ComBat [Citation34], included in the SVA package [Citation35]. Next, we used the SVA package to capture technical covariates to be included in our linear regression model, and included the recommended one surrogate variable in our model. Next, we used multivariable linear regression contained within the limma R package to measure differential DNAm on the β values. Since our study design has a very small sample size (N = 3 control, N = 3 CS) and our p values would therefore not accurately reflect very small differences [Citation36] in DNAm, we considered CpGs as significant at p < 10e-3 and an effect size >0.05. We produced figures in R using the ggplot2 and Gviz packages.
Gene ontology
To obtain biological significance from identified differentially methylated probes, we used the ClusterProfiler R package [Citation37] (v3.16.1) for gene enrichment analysis. We considered pathways with FDR < 0.05 to be significantly enriched.
Results
The research presented here is one part of a larger animal experiment to investigate the effects of prenatal CS exposure on offspring. We exposed BALB/c dams (N = 16 controls and N = 16 CS-exposed) to whole-body CS for a total period of 9 weeks, starting 3 weeks before mating, and lasting throughout pregnancy until 3 weeks after the birth of their pups (). CS-exposed dams tolerated smoking, with no losses or adverse events during the course of the experiments. We performed lung function and collected tissues for further analyses at two timepoints: 48–72 hours after the last day of 9 weeks of smoking (referred to hereafter as ‘immediately after smoking’), and 15–16 weeks after smoking cessation (referred to hereafter as ‘15 weeks after smoking cessation’) (, Supplementary Table S1).
Figure 1. Development of a mouse model to study the effects of heavy smoking. (a) Adult female mice (N = 16 control, N = 16 CS) were exposed to heavy doses of whole-body CS for 9 weeks, from a pre-pregnancy period of 3 weeks and ending 3 weeks after birth of offspring. 15 of these had their litters within one week of each other and were included in this study. Following lung function measurements, tissues were collected from dams 48 hours after the last day of 9 weeks of smoke exposure and then 15 weeks after smoking cessation. (b) Dam cotinine levels measured in plasma 48 hours and 15 weeks after smoking cessation. N = 5 per group. Differences in plasma cotinine were analysed using student t-tests.
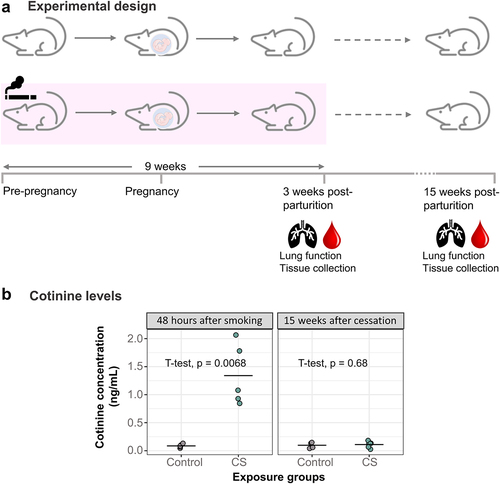
To verify absorption of CS components, we measured cotinine levels in plasma collected at these 2 different timepoints (). Dams which were exposed to CS had 15 times more elevated plasma cotinine levels (Mean = 1.34, SD = 0.55) immediately after smoking cessation compared to control dams (Mean = 0.09, SD = 0.03) (, p = 0.0068). The relatively low levels of cotinine observed overall in the smoke-exposed dams can be attributed to the collection of plasma from dams approximately 48 hours after smoking, with the reported half-life of cotinine being about 24 hours [Citation38,Citation39]. Cotinine levels measured 15 weeks after CS exposure showed no significant difference between control and smoking dams ().
Lung phenotype immediately after 9 weeks of smoking and 15 weeks after smoking cessation
Several studies have shown that personal smoking alters lung function and induces airway hyperresponsiveness [Citation40–42]. Therefore, we assessed the effects of chronic smoking on the lung phenotype by measuring lung function and immune cell infiltration.
Smoking for 9 weeks caused a fourfold and eightfold increase in eosinophils and lymphocytes, respectively, immediately after smoking, compared to controls (). The immune cell infiltration differences, however, did not persist until 15 weeks after smoking cessation (). Also of note is that total cell count and macrophage cell count were higher in both groups at the immediate post-smoking timepoint, when the dams were 3 weeks post-parturition and still lactating, than 15 weeks later (). We also found that direct CS exposure did not alter dam lung function either at baseline () or after methacholine challenge after 9 weeks of smoking (), though tissue elastance was lower but not statistically significant in CS exposed than unexposed dams (). Thirteen weeks after smoking cessation, baseline lung function was also not altered (), but we observed significant differences between CS exposed and unexposed animals in tissue elastance, tissue resistance and total lung resistance values ().
Figure 2. Smoke exposure causes transient changes to immune cell infiltration into dam lungs, but does not alter dam baseline lung function over time. (a) Total immune cells per mL lavage immediately after smoking and 15 weeks after smoking cessation. (b) Eosinophils per mL lavage in the CS-exposed dams (mean = 7528, SD = 3736) was over 4 times more elevated than control dams (mean = 1569, SD = 1029) immediately after smoking, but normalized to controls 15 weeks after smoking cessation. (c) Macrophages per mL lavage immediately after smoking and 15 weeks after smoking cessation. (d) Lymphocytes per mL lavage in the CS-exposed dams (mean = 5680, SD = 2357) was over 8 times elevated compared to control dams (mean = 649, SD = 256) immediately after smoking, but normalized to controls 15 weeks after smoking cessation. (e) Total lung resistance at baseline. (f) Airway resistance at baseline. (g) Tissue resistance at baseline. (h) Alveolar elastance at baseline. N = 4–6 per group. Differential cell counts were normalized to lavage volume. Lung function values were measured using 90th percentile values after injection of saline into the lungs. Two-group comparisons were conducted using a student t-test and p < 0.05 was considered significant.
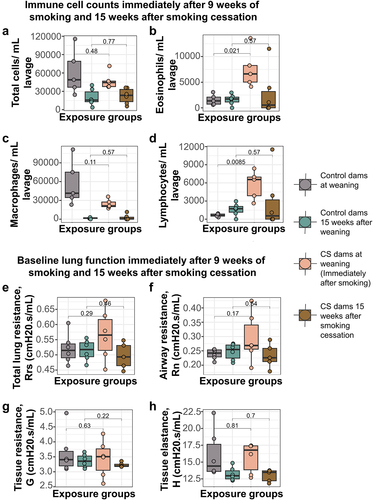
Figure 3. Smoke exposure alters methacholine responsiveness in mouse lungs up to 15 weeks after smoking cessation. (a) Total lung resistance immediately after smoking. (b) Airway resistance immediately after smoking. (c) Tissue resistance immediately after smoking. (d) Alveolar elastance immediately after smoking. (e) Total lung resistance after 15 weeks of smoking cessation. (f) Airway resistance after 15 weeks of smoking cessation. (g) Tissue resistance after 15 weeks of smoking cessation. (h) Alveolar elastance after 15 weeks of smoking cessation. N = 5–6 per group. Lung function values were measured using 90th percentile values upon administration of increasing doses of methacholine. Data was analyzed using one-way ANOVA, followed by multiple comparisons at each methacholine dose where significant. *p < 0.05 in control vs smoke-exposed group.
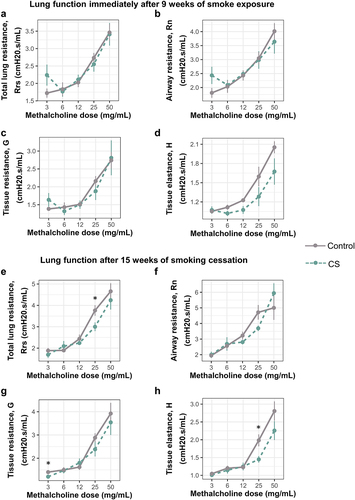
Together, these data show that smoking for 9 weeks alters lung phenotype by increasing immune cell infiltration into the lungs immediately after smoking, accompanied by persistent decline in lung function even after smoking cessation.
Candidate gene DNAm and expression immediately after 9 weeks of smoking and 15 weeks after smoking cessation
To determine whether CS-induced DNAm changes observed in humans are recapitulated in our mouse model, we identified candidate genes to assess initially via pyrosequencing. We first measured DNAm and expression at Cyp1a1 immediately after smoking and 15 weeks after cessation. We found an increase in Cyp1a1 DNAm in the blood of CS-exposed dams 15 weeks after smoking cessation compared to controls, though this increase was not statistically significant (). Cyp1a1 expression in the lungs of CS-exposed dams was slightly but not significantly elevated 15 weeks after smoking cessation (). Chronic exposure to CS for 9 weeks led to a significant decrease in Cyp1a1 DNAm in the left lungs of CS-exposed dams immediately after smoking (, Mean = 11.20, SD = 1.05), compared to controls (, Mean = 19.20, SD = 0.80). Interestingly, Cyp1a1 DNAm remained significantly decreased in the lungs of smoking dams (, Mean = 15.00, SD = 1.07) compared to controls (, Mean = 17.60, SD = 0.58) till 15 weeks after smoking cessation, though the magnitude had reduced slightly.
Figure 4. Cyp1a1 and Ahrr DNAm and expression levels in dam blood and lungs immediately after smoking and 15 weeks after smoking cessation. (a) Cyp1a1 DNAm in dam blood 15 weeks after smoking cessation. (b) Cyp1a1 expression in dam lungs 15 weeks after smoking cessation. (c) Cyp1a1 DNAm in dam lungs immediately after 9 weeks of smoking (d) Cyp1a1 DNAm in dam lungs 15 weeks after smoking cessation. (e) Ahrr DNAm in dam blood after 15 weeks of smoking cessation. (f) Ahrr DNAm in dam lungs after 15 weeks of smoking cessation. (g) Ahrr expression in dam lungs after 15 weeks of smoking cessation. N = 2–5 per group. Differences in DNAm and expression were analysed using student t-tests.
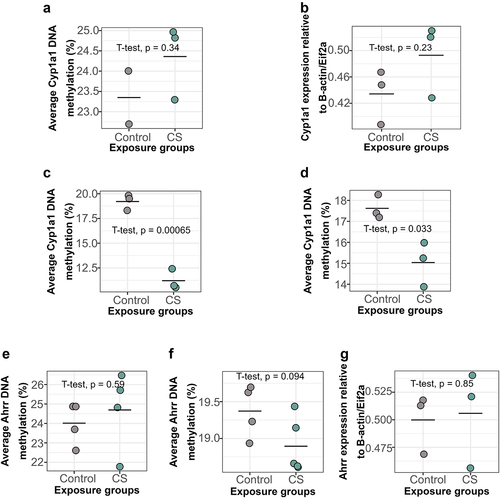
Conversely, we found no significant differences in Ahrr DNAm in dam blood 15 weeks after smoking cessation (). Similarly, Ahrr DNAm () and Ahrr expression () in dam lungs at this same timepoint was also not significantly different between groups. These results thus show that personal smoking for 9 weeks causes alterations in Cyp1a1 DNAm in the lungs which persist for 15 weeks after smoking cessation.
Measurement of effects of smoking on mouse lungs across the whole mouse epigenome using the Illumina mouse methylation microarrays
Following candidate gene DNAm measurements at Ahrr and Cyp1a1, we evaluated the association between smoking and epigenome-wide DNAm in the lungs immediately after 9 weeks of smoking, using normalized β values from the Illumina mouse microarrays. Due to our small sample size (N = 2–6 per group), we used a combination of p value (p < 1e-3) and effect size (0.05) cut-offs to determine significance. After adjusting for the one surrogate variable recommended by SVA (Supplementary Figure S1(a)), we performed multivariable regression on beta values from the autosomal probes (Supplementary Figure S1(a)) and identified 40 significant CpGs differentially methylated in dams exposed to heavy doses of CS for 9 weeks (). Maternal smoking in pregnancy led to higher DNAm at 37.5% (15 CpGs) and lower DNAm at 62.5% (25 CpGs) of these significant CpGs (). While our candidate gene, Cyp1a1, was not among our list of significant genes, a closer look at each of the Cyp1a1 probes on the mouse array reveal large differences in DNAm between controls and CS-exposed dams in 3 of 9 Cyp1a1 probes that passed preprocessing (Supplementary Figure S2, positions E, F and G). The second candidate gene, Ahrr, was also not significant in our epigenome-wide analysis and showed small effect size differences overall (Supplementary Figure S3).
Table 1. CpGs significantly altered immediately after exposure to CS for 9 weeks. Linear regression was computed using LIMMA, after adjusting for one surrogate variable recommended by SVA (see Figure S1).
We performed a similar analysis on probes from the X chromosome, and after adjusting for the 1 surrogate variable recommended by SVA, found no significantly altered CpGs in dam lungs immediately after smoking (Supplementary Figure S4).
Gene Ontology terms associated with smoking in the lungs
To gain insights into biological processes potentially affected by smoking in the lungs, we performed gene ontology analyses on genes mapping to the identified 40 significant CpGs. Gene ontology analysis revealed 54 biological processes which passed the FDR cut-off of 0.05 (). The top pathway findings were mostly driven by Sox11, Wnt5a, Npnt, Slit3, Magi2 and Furin, all being involved in numerous processes. The most significantly implicated pathway with the smallest FDR (FDR = 0.007) was involved in regulation of transmembrane receptor protein serine/threonine kinase signalling pathways.
Table 2. List of significantly enriched biological pathways/processes immediately after 9 weeks of smoking.
Discussion
Cigarette smoke is a complex mixture of over 7000 components [Citation43–46], which affect virtually all body systems via mechanisms such as inflammation and DNA damage. While the processes linking smoking and adverse health outcomes are poorly understood, DNA methylation alterations have been identified as possible links due to their sensitivity to the environment and relative stability [Citation47]. The association between smoking and changes in DNAm has been well documented, especially in human blood [Citation6–9]. However, while there may be shared DNAm patterns between tissues [Citation14,Citation18], overall DNAm is highly variable across tissues [Citation15–17], emphasizing the need to study smoke-induced DNAm alterations in more directly affected tissues such as the lungs. The major aim of this study, therefore, was to identify DNAm changes in mouse lungs arising due to smoking, and determine how these patterns would change after a period of smoking cessation. To this effect, we adapted an effective smoking mouse model which we have also used to measure the effects of early life smoke exposure on offspring [Citation23]. Using this model, we identified 40 lung-specific CpGs which were differentially methylated following chronic smoke exposure, 32 of which had never been linked to smoking in the past. We also showed that chronic smoking causes alterations in mouse lung function that can last into adulthood, even after a long period of smoking cessation. These results are important, as identification of lung-specific DNAm patterns may provide more effective biomarkers for smoking, and may potentially be used to develop therapeutic interventions against the effects of chronic smoke exposure.
Prior to this study, much of what was known about smoking and DNAm was from studies conducted in blood in humans. Only a quarter of the sites we identified were associated with genes that had previously been connected to CS exposure in any tissue, and of those, there was existing evidence about DNAm differences specifically only at one gene, Fkbp5 [Citation48]. One study reported that smoking decreases Fkbp5 DNAm in blood, which might be connected to dysregulation of the hypothalamic–pituitary–adrenal axis [Citation48]. Our study replicated this finding, as we also observed a decrease in Fkbp5 DNAm in mouse lungs our study, which might implicate the hypothalamic–pituitary–adrenal axis more broadly across tissues in the response to early life CS exposure.
Of the other genes with prior evidence of connection to CS exposure, the majority have genetic or transcriptomic connections. For example, one of our hits mapped to the Ataxin two (Atxn2) gene, expression of which has been linked to pancreatic cancer development in smokers [Citation49]. The CpG we identified is located in an intragenic exon which is actively transcribed in mouse lungs, with high levels of H3K36me3 [Citation50–52]. Generally, the function of DNAm at exons varies depending on where the exon is located [Citation53–55], but most studies have discovered positive correlations between DNAm in intergenic exons and expression levels [Citation53,Citation56]. As our significant CpG is located in an intragenic exon, increased DNAm in smoking dams could signal an increase in Atxn2 expression linked with an increased risk of malignancy. This is the first study associating smoking with altered DNAm at Atxn2.
We also found for the first time that heavy smoking resulted in a significant decrease in lung DNAm at Dopa decarboxylase (Ddc), a gene encoding a protein which catalyses the final steps of dopamine and serotonin biosynthesis [Citation57]. Genetic variants in Ddc, especially at the introns [Citation58], have been associated with smoking behaviour and nicotine dependency [Citation58–60] in African-American and European-American populations. It therefore follows that alterations in DNAm at this gene as observed here could contribute to nicotine dependency in individuals who smoke. It is however important to note that genetic variants or genes alone do not determine susceptibility to addiction, and that the environment plays a major role [Citation61,Citation62]. In a Finnish twin study, it was discovered that the influence of genetics in adolescent smokers was decreased when the adolescents were highly monitored by their parents [Citation63]. This emphasizes the need for more research to focus on understanding gene–environment interactions, as it could be crucial in deciphering the mechanism behind addiction.
The six other genes with prior evidence for association with smoking are summarized as follows: 1) Slit guidance ligand three (Slit3) whose expression has been associated with the development of nicotine preference in zebrafish [Citation64], and which is down-regulated in human lung adenocarcinoma cell lines and tissues [Citation65]. Expression levels of SLIT3 are also significantly correlated with smoking history, as non-smokers with high expression of SLIT3 had better prognosis among lung adenocarcinoma patients [Citation65]; 2) Wingless-type MMTV integration site family, member 5A (Wnt5a) which is upregulated by tobacco smoke condensate in lung cancer cells [Citation66], and in mouse and human lung tissues [Citation67], and has been associated with the development of lung carcinogenesis in bronchial epithelial cells of smokers [Citation68]; 3) Ankyrin repeat and SOCS box-containing 18 (Asb18) which shows differential expression levels in smokers [Citation69]; 4) Nephronectin (Npnt), whose variants were shown to be involved in COPD-mediated airflow obstruction both in heavy smokers and non-smokers in a UK-based cohort [Citation70]; 5) Membrane associated guanylate kinase, WW and PDZ domain containing 2 (Magi2) which shows higher expression in bronchial biopsies of heavy smokers with COPD [Citation71]. Magi2 polymorphisms may also influence nicotine dependence in smokers [Citation72]; and 6) FK506 binding protein 5 (Fkbp5) which has been linked to DNAm changes in blood as discussed, but also modulates the effects of nicotine on the hypothalamic pituitary adrenal axis in female smokers [Citation73].
Of the 32 novel sites identified in this study, two mapped to the promoter of B3gnt3 (UDP-GlcNAc:betaGal beta-1,3-N-acetylglucosaminyltransferase 3) gene. Both CpGs, which are 32 bp apart, showed significantly higher DNAm in the lungs of dams exposed to CS compared to controls. This gene codes for a type II transmembrane protein which is involved in the biosynthesis of poly-N-acetyllactosamine chains, L-selectin ligand biosynthesis, lymphocyte homing and lymphocyte trafficking and recirculation [Citation74,Citation75]. One study reported increased B3gnt3 expression levels corresponding to increased immune cell infiltration and correlated with poorer outcomes in patients with lung adenocarcinoma [Citation76]. Other studies have also associated differential B3gnt3 expression levels with development of many other types of cancers [Citation77–81]. It therefore follows that increased DNAm in our two CpG sites, which are located in a weak promoter, could translate to decreased expression of the gene, consequently resulting in increased tolerance to and reduced sensitivity and response to foreign antigens by T cells. This is the first study associating smoking with altered DNAm at B3gnt3.
While there is a relative dearth of studies examining DNAm in the lungs, we found three other studies which directly measured effects of personal smoking on DNAm in the lungs. The first measured epigenome-wide DNAm in the blood of current and never-smokers. Next, using the identified significant CpGs from the epigenome-wide analysis, they measured DNAm in the lungs of current, ex- and never-smokers using pyrosequencing [Citation14]. Out of 15 CpGs differentially methylated sites in the blood, only five replicated in the lungs, confirming that while these tissues share some similarities, not all blood-based findings will replicate in the lung [Citation14]. Two of their five CpGs mapped to AHRR, and one corresponded to the human position we used to identify our mouse candidate Ahrr position (cg05575921). We did not observe DNAm changes in the lungs at Ahrr, but its poor conservation with the human genome in this region may limit comparability. In the second study, the effects of a) whole-body CS, b) the tobacco carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, and c) the inflammatory agent lipopolysaccharide were measured in mouse lungs using RRBS [Citation20]. They found DNAm and hydroxymethylation differences across multiple differentially methylated regions [Citation20]. While there were no overlapping differentially methylated sites between their results and ours, they identified 10 hydroxymethylated sites which corresponded to, or whose isoforms corresponded to 10 of our differentially methylated sites: Strip1, Slit3, Megf6, Magi2, Atxn2, Sox10/Sox18, Wnt5b, Fkbp3, Asb13 and Fbxo41/Fbxo45. The microarrays we used here cannot distinguish between methylcytosine and hydroxymethylcytosine, and so it is possible that some of our findings are due to similar hydroxymethylcytosine changes. A third study exposed mice to 2 hours of CS twice daily, for 4 weeks consecutively in order to analyse the effect of smoking on lung DNAm. Using liquid hybridization capture-based bisulphite sequencing, they found that smoking altered DNAm patterns at numerous genes, most of which were involved in inflammation and in inflammatory injury in COPD, but due to limits on data availability it was not possible to determine whether specific findings overlapped with ours [Citation21].
Gene ontology analysis on our list of significant genes revealed enrichment in multiple biological processes, with the most significant being involved in regulation of transmembrane receptor protein serine/threonine kinase signalling pathways. Components of CS, such as nicotine and the CS-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, when bound to nicotinic-acetylcholine receptors, activate serine/threonine kinase Akt [Citation82]. Activation of Akt resulted in increased phosphorylation of downstream molecules involved in controlling the cell cycle and protein translation, such as glycogen synthase kinase 3 (GSK-3) [Citation82]. This in turn leads to cancer-like phenotypes such as loss of contact inhibition, desensitization to regulation by growth factors, decreased apoptosis, angiogenesis and increased cellular proliferation [Citation82,Citation83]. Since our current study identified an enrichment for genes involved in regulation of serine/threonine Akt pathways, it could support a predisposition to lung tumorigenesis in smoke-exposed mice.
Our candidate gene DNAm analysis also revealed interesting patterns. Our first candidate gene, AHRR, particularly locus cg05575921, has repeatedly shown decreased DNAm in human serum [Citation84], whole and peripheral blood [Citation9,Citation85], saliva [Citation86], monocytes [Citation87] and alveolar macrophages [Citation10] of current smokers compared to never-smokers. In the same vein, even offspring exposed to CS in utero have shown altered AHRR DNAm at the same locus compared to controls [Citation28,Citation88–90]. Compared to these studies however, our candidate gene analysis did not reveal significant differences in Ahrr DNAm or expression in blood or lungs at the corresponding mouse locus. This was interesting but not entirely surprising, since Ahrr is not well conserved between mice and humans at this region. Therefore, further mapping of DNAm in the Ahrr gene in mouse lung may reveal deeper insights.
At Cyp1a1, we found that smoking for 9 weeks caused a significant decrease in DNAm immediately after smoking, and that this pattern remained significant until 15 weeks after smoking cessation. However, Cyp1a1 expression in mouse lungs after smoking cessation, though increased, was not significant, indicating that DNAm changes may last after expression differences have subsided. Cyp1a1 belongs to the cytochrome p450 enzyme subclass, and is involved in drug/xenobiotic metabolism [Citation91–93]. Therefore, reduced DNAm of Cyp1a1 in CS-exposed mice as observed in this study could indicate induction of the enzyme to aid detoxification of CS components. These results mimic reduced Cyp1a1 DNAm and increased expression patterns observed in the lungs [Citation94], adipose tissue [Citation95], prostate cancer tissues [Citation96], buccal cells [Citation19] and blood [Citation19] of humans exposed to CS in previous studies. Even though none of the Cyp1a1 probes in the mouse methylation microarray was among our 40 significant CpGs, we showed that 3 of the 9 Cyp1a1 probes have large differences in DNAm. Our inability to detect these positions (and likely some other CpGs) as significant is likely due to our small sample size. So, while these 3 probes met our strict effect size cut-off of 0.05, they did not meet the p value cut-off. Future studies with larger sample sizes would be helpful in generating more precise measurements.
In addition to molecular alterations, we also assessed the effects of chronic smoking on physiological outcomes in the form of lung function. We observed that the changes in lung function immediately after smoking for 9 weeks were small and not significant. However, 15 weeks after smoking cessation, lung function in CS-exposed dams had significantly declined, as evidenced by increased airway responsiveness to methacholine. One reason why lung function worsens over time in our smoke-exposed mice may be due to ageing. Past research indicates that lung function declines with age due to structural and immunological factors which impair gas exchange and increase susceptibility to infections [Citation97–99]. Other studies report that smoking exacerbates this lung function decline, even in ex-smokers [Citation100–103]. This trend of worsening/decline in lung function is a characteristic of COPD [Citation104,Citation105]. In line with our finding of declining lung function even with smoking cessation, in a large multi-ethnic pooled cohort study [Citation103], smoking cessation slightly improved lung function, but these ‘improvements’ still resulted in lung function similar to light/low-intensity current smokers. In fact, smoking cessation does not completely restore lung function to the levels of never-smokers [Citation103]. This pattern of lung function decline despite smoking cessation may be due to sustained dysregulation of epigenetic patterns [Citation106–108], immune responses [Citation109–111] and airway hyperresponsiveness [Citation112].
This study represents an important contribution to our understanding of the impacts of CS on the lungs, but is not without limitations. The first limitation is our small sample size for the EWAS and hence, low statistical power in identifying small DNAm differences. We also could not distinguish between hydroxymethylcytosine and methylcytosine, meaning that some of our differential DNAm findings may be due to changes in hydroxymethylcytosine. It is also worth noting that whole lungs were used in this study, making it impossible to determine whether differences in cell composition are masking some of our findings. However, we took two approaches to mitigate this problem. First, we lavaged the lungs prior to analysis to remove immune cells which we know to be different between CS and control animals, making it unlikely that immune cells are responsible for our observed results. To further support this, we found that Cyp1a1 DNAm in the blood of adult female control mice () is higher than Cyp1a1 DNAm in lungs of control mice (); therefore, infiltration of immune cells from blood to lungs is unlikely to cause the decrease in lung Cyp1a1 DNAm in the present study. Second, we used surrogate variables to control for variance likely associated with cell type. This approach removes the effects of cell type such that further work will be required to determine which lung cell types exhibit the observed DNAm differences.
In summary, we have shown here that chronic smoking alters DNAm patterns in mouse lungs at multiple CpGs, some which have never been associated with smoking in the past. These CpGs map to genes implicated in tumorigenesis and metastasis, neurodegenerative disorders, nicotine dependency and immune cell responses. Moreover, lung function defects persist up to 15 weeks after smoking cessation. The fact that 75% of the significant CpGs reported here have never been linked to CS in the past, even in studies which investigated effects of CS in the blood, emphasizes the need for more studies to be done on the lungs, as it clearly has different responses than other tissues. This research is one part of a much larger study to investigate the effects of early life CS exposure on offspring DNAm patterns. Therefore, the results presented here will hopefully also shed more light on some of the patterns we observe in offspring exposed to CS in utero.
Figure S4_dam paper.tif
Download TIFF Image (2.2 MB)Table S1_Sample information.xlsx
Download MS Excel (10.2 KB)Figure S2_Cyp1a1_Gviz_dam paper.tif
Download TIFF Image (2 MB)Figure S3_dam paper.tif
Download TIFF Image (1.7 MB)Figure S1_dam paper.tif
Download TIFF Image (1.9 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15592294.2024.2322386
Data availability statement
The data supporting the findings from this study are available from the corresponding author, M.J.J., upon request.
Additional information
Funding
References
- Tamimi A, Serdarevic D, Hanania NA. The effects of cigarette smoke on airway inflammation in asthma and COPD: therapeutic implications. Respir med. 2012;106(3):319–20. doi: 10.1016/j.rmed.2011.11.003
- Boulet L-P, Catherine L, Francine A, et al. Smoking and asthma: clinical and radiologic features, lung function, and airway inflammation. Chest. 2006;129(3):661–668. doi: 10.1378/chest.129.3.661
- Nakano Y, MURO S, SAKAI H, et al. Computed tomographic measurements of airway dimensions and emphysema in smokers. Am J Respir Crit Care Med. 2000;162(3):1102–1108. doi: 10.1164/ajrccm.162.3.9907120
- Pietinalho A, Pelkonen A, Rytilä P. Linkage between smoking and asthma. Allergy. 2009;64(12):1722–1727. doi: 10.1111/j.1398-9995.2009.02208.x
- Piipari R. Smoking and asthma in adults. Eur Respir J. 2004;24(5):734–739. doi: 10.1183/09031936.04.00116903
- Elliott HR, Tillin T, McArdle WL, et al. Differences in smoking associated DNA methylation patterns in South Asians and Europeans. Clin Epigenetics. 2014;6(1). doi: 10.1186/1868-7083-6-4
- Su D, Wang X, Campbell MR, et al. Distinct epigenetic effects of tobacco smoking in whole blood and among leukocyte subtypes. PLoS One. 2016;11(12):e0166486. doi: 10.1371/journal.pone.0166486
- Zhang Y, Florath I, Saum K-U, et al. Self-reported smoking, serum cotinine, and blood DNA methylation. Environ Res. 2016;146:395–403. doi:10.1016/j.envres.2016.01.026
- Zeilinger S, Kühnel B, Klopp N, et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS One. 2013;8(5):e63812. doi: 10.1371/journal.pone.0063812
- Monick MM, Beach SRH, Plume J, et al. Coordinated changes in AHRR methylation in lymphoblasts and pulmonary macrophages from smokers. Am J Med Genetics Pt B. 2012;159B(2):141–151. doi: 10.1002/ajmg.b.32021
- Oliveira NFP, Damm GR, Andia DC, et al. DNA methylation status of the IL8 gene promoter in oral cells of smokers and non-smokers with chronic periodontitis. J Clinic Periodontol. 2009;36(9):719–725. doi: 10.1111/j.1600-051X.2009.01446.x
- Bollepalli S, Korhonen T, Kaprio J, et al. EpiSmokEr: a robust classifier to determine smoking status from DNA methylation data. Epigenomics. 2019;11(13):1469–1486. doi: 10.2217/epi-2019-0206
- Zhang Y, Elgizouli M, Schöttker B, et al. Smoking-associated DNA methylation markers predict lung cancer incidence. Clin Epigenetics. 2016;8(1):127. doi: 10.1186/s13148-016-0292-4
- de Vries M, van der Plaat DA, Nedeljkovic I, et al. From blood to lung tissue: effect of cigarette smoke on DNA methylation and lung function. Respir Res. 2018;19(1):212. doi: 10.1186/s12931-018-0904-y
- Bloushtain-Qimron N,Yao J, Snyder EL et al. Cell type-specific DNA methylation patterns in the human breast. Proceedings of the National Academy of Sciences. 2008; 105, 14076–14081.
- Zhou J, Sears RL, Xing X, et al. Tissue-specific DNA methylation is conserved across human, mouse, and rat, and driven by primary sequence conservation. BMC Genomics. 2017;18(1):724. doi: 10.1186/s12864-017-4115-6
- Scherer M, Gasparoni G, Rahmouni S, et al. Identification of tissue-specific and common methylation quantitative trait loci in healthy individuals using MAGAR. Epigenet Chromatin. 2021;14(1):44. doi: 10.1186/s13072-021-00415-6
- Stueve TR, Li W-Q, Shi J, et al. Epigenome-wide analysis of DNA methylation in lung tissue shows concordance with blood studies and identifies tobacco smoke-inducible enhancers. Hum Mol Genet. 2017;26(15):3014–3027. doi: 10.1093/hmg/ddx188
- Teschendorff AE, Yang Z, Wong A, et al. Correlation of smoking-associated DNA methylation changes in buccal cells with DNA methylation changes in epithelial cancer. JAMA Oncol. 2015;1(4):476–485. doi: 10.1001/jamaoncol.2015.1053
- Seiler CL, Song JUM, Kotandeniya D, et al. Inhalation exposure to cigarette smoke and inflammatory agents induces epigenetic changes in the lung. Sci Rep. 2020;10(1):11290. doi: 10.1038/s41598-020-67502-8
- Li P, Peng J, Chen G, et al. DNA methylation profiling in a cigarette smoke-exposed mouse model of airway inflammation. J Chronic Obstructive Pulmonary Dis. 2022;17:2443–2450. doi: 10.2147/COPD.S369702
- Zhou W, Hinoue T, Barnes B, et al. DNA methylation dynamics and dysregulation delineated by high-throughput profiling in the mouse. Cell Genom. 2022;2(7):100144. doi: 10.1016/j.xgen.2022.100144
- Onuzulu CD, Lee S, Basu S, et al. Early-life exposure to cigarette smoke primes lung function and DNA methylation changes at Cyp1a1 upon exposure later in life. Am J Physiol Lung Cell Mol Physiol. 2023;325(5):L552–L567. doi: 10.1152/ajplung.00192.2023
- Ma Y, Halayko AJ, Basu S, et al. Sustained suppression of IL-13 by a vaccine attenuates airway inflammation and remodeling in mice. Am J Respir Cell Mol Biol. 2013;48(5):540–549. doi: 10.1165/rcmb.2012-0060OC
- Ryu MH, Jha A, Ojo OO, et al. Chronic exposure to perfluorinated compounds: impact on airway hyperresponsiveness and inflammation. Am J Physiol Lung Cell Mol Physiol. 2014;307(10):L765–L774. doi: 10.1152/ajplung.00100.2014
- ElDin NB, El-Rahman MKA, Zaazaa HE, et al. Microfabricated potentiometric sensor for personalized methacholine challenge tests during the COVID-19 pandemic. Biosens Bioelectron. 2021;190:113439. doi:10.1016/j.bios.2021.113439
- Coates AL, Wanger J, Cockcroft DW, et al. ERS technical standard on bronchial challenge testing: general considerations and performance of methacholine challenge tests. Eur Respir J. 2017;49(5):1601526. doi: 10.1183/13993003.01526-2016
- Joubert BR, Felix J, Yousefi P, et al. DNA methylation in newborns and maternal smoking in pregnancy: genome-wide consortium meta-analysis. Am J Hum Genet. 2016;98(4):680–696. doi: 10.1016/j.ajhg.2016.02.019
- MUSCLE: multiple sequence alignment with improved accuracy and speed | IEEE Conference Publication | IEEE Xplore. Available from: https://ieeexplore.ieee.org/abstract/document/1332560
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262
- Wang R, Yan B, Li Z, et al. Long non‑coding RNA HOX transcript antisense RNA promotes expression of 14‑3‑3σ in non‑small cell lung cancer. Exp Ther Med. 2017;14:4503–4508. doi: 10.3892/etm.2017.5041
- Zhou W, Triche TJ, Laird PW, et al. SeSAMe: reducing artifactual detection of DNA methylation by Infinium BeadChips in genomic deletions. Nucleic Acids Res. 2018;19:129. doi:10.1093/nar/gky691
- Triche TJ, Weisenberger DJ, Van Den Berg D, et al. Low-level processing of Illumina infinium DNA methylation BeadArrays. Nucleic Acids Res. 2013;41(7):e90–e90. doi: 10.1093/nar/gkt090
- Johnson WE, Li C, Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8(1):118–127. doi: 10.1093/biostatistics/kxj037
- Leek JT, Storey JD, Gibson G. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007;3(9):e161. doi: 10.1371/journal.pgen.0030161
- Gómez-de-Mariscal E, Guerrero V, Sneider A, et al. Use of the p-values as a size-dependent function to address practical differences when analyzing large datasets. Sci Rep. 2021;11(1):20942. doi: 10.1038/s41598-021-00199-5
- Wu T, Hu E, Xu S, et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation. 2021;2(3):100141. doi: 10.1016/j.xinn.2021.100141
- Leong JW, Dore ND, Shelley K, et al. The elimination half-life of urinary cotinine in children of tobacco-smoking mothers. Pulm Pharmacol Ther. 1998;11(4):287–290. doi: 10.1006/pupt.1998.0153
- Benowitz NL. Cotinine as a biomarker of environmental tobacco smoke exposure. Epidemiol Rev. 1996;18(2):188–204. doi: 10.1093/oxfordjournals.epirev.a017925
- Melgert BN, Postma DS, Geerlings M, et al. Short-term smoke exposure attenuates ovalbumin-induced airway inflammation in allergic mice. Am J Respir Cell Mol Biol. 2004;30(6):880–885. doi: 10.1165/rcmb.2003-0178OC
- Blacquière MJ, Timens W, Melgert BN, et al. Maternal smoking during pregnancy induces airway remodelling in mice offspring. Eur Respir J. 2009;33(5):1133–1140. doi: 10.1183/09031936.00129608
- Lei Y, Cao Y-X, Xu C-B, et al. The raf-1 inhibitor GW5074 and dexamethasone suppress sidestream smoke-induced airway hyperresponsiveness in mice. Respir Res. 2008;9(1):71. doi: 10.1186/1465-9921-9-71
- Colombo G, Dalle-Donne I, Orioli M, et al. Oxidative damage in human gingival fibroblasts exposed to cigarette smoke. Free Radic Biol Med. 2012;52(9):1584–1596. doi: 10.1016/j.freeradbiomed.2012.02.030
- Zeng T, Liu Y, Jiang Y, et al. Advanced materials design for adsorption of toxic substances in cigarette smoke. Adv Sci. 2023;10(22):2301834. doi: 10.1002/advs.202301834
- Soleimani F, Dobaradaran S, De-la-Torre GE, et al. Content of toxic components of cigarette, cigarette smoke vs cigarette butts: a comprehensive systematic review. Sci Total Environ. 2022;813:152667. doi:10.1016/j.scitotenv.2021.152667
- Staal YCM, Bos PMJ, Talhout R. Methodological approaches for risk assessment of tobacco and related products. Toxics. 2022;10(9):491. doi: 10.3390/toxics10090491
- Nwanaji-Enwerem JC, Colicino E. DNA methylation–based biomarkers of environmental exposures for human population studies. Curr Envir Health Rpt. 2020;7(2):121–128. doi: 10.1007/s40572-020-00269-2
- Dogan MV, Lei M-K, Beach SRH, et al. Alcohol and tobacco consumption alter hypothalamic pituitary adrenal axis DNA methylation. Psychoneuroendocrinology. 2016;66:176–184. doi:10.1016/j.psyneuen.2016.01.018
- Tang H, Wei P, Duell EJ, et al. Axonal guidance signaling pathway interacting with smoking in modifying the risk of pancreatic cancer: a gene- and pathway-based interaction analysis of GWAS data. Carcinogenesis. 2014;35(5):1039–1045. doi: 10.1093/carcin/bgu010
- Gorkin DU, Barozzi I, Zhao Y, et al. An atlas of dynamic chromatin landscapes in mouse fetal development. Nature. 2020;583(7818):744–751. doi: 10.1038/s41586-020-2093-3
- Ernst J, Kellis M. ChromHMM: automating chromatin state discovery and characterization. Nat Methods. 2012;9(3):215–216. doi: 10.1038/nmeth.1906
- Sloan CA, Chan ET, Davidson JM, et al. ENCODE data at the ENCODE portal. Nucleic Acids Res. 2016;44(D1):D726–32. doi: 10.1093/nar/gkv1160
- Li S, Zhang J, Huang S, et al. Genome-wide analysis reveals that exon methylation facilitates its selective usage in the human transcriptome. Brief Bioinform. 2018;19(5):754–764. doi: 10.1093/bib/bbx019
- Song K, Li L, Zhang G, et al. The association between DNA methylation and exon expression in the Pacific oyster crassostrea gigas. PLoS One. 2017;12(9):e0185224. doi: 10.1371/journal.pone.0185224
- Brenet F, Moh M, Funk P, et al. DNA methylation of the first exon is tightly linked to transcriptional silencing. PLoS One. 2011;6(1):e14524. doi: 10.1371/journal.pone.0014524
- Chuang T-J, Chen F-C, Chen Y-Z. Position-dependent correlations between DNA methylation and the evolutionary rates of mammalian coding exons. Proc Natl Acad Sci U S A. 2012;109(39):15841–15846. doi: 10.1073/pnas.1208214109
- Ng J, Papandreou A, Heales SJ, et al. Monoamine neurotransmitter disorders—clinical advances and future perspectives. Nat Rev Neurol. 2015;11(10):567–584. doi: 10.1038/nrneurol.2015.172
- Yu Y, Panhuysen C, Kranzler HR, et al. Intronic variants in the dopa decarboxylase (DDC) gene are associated with smoking behavior in European-Americans and African-Americans. Hum Mol Genet. 2006;15(14):2192–2199. doi: 10.1093/hmg/ddl144
- Ma JZ, Beuten J, Payne TJ, et al. Haplotype analysis indicates an association between the DOPA decarboxylase (DDC) gene and nicotine dependence. Hum Mol Genet. 2005;14(12):1691–1698. doi: 10.1093/hmg/ddi177
- Zhang H, Ye Y, Wang X, et al. DOPA decarboxylase gene is associated with nicotine dependence. Pharmacogenomics. 2006;7(8):1159–1166. doi: 10.2217/14622416.7.8.1159
- Kirst M, Mecredy G, Borland T, et al. Predictors of substance use among young adults transitioning away from high school: a narrative review. Substance Use Misuse. 2014;49(13):1795–1807. doi: 10.3109/10826084.2014.933240
- Vink JM. Genetics of addiction: future focus on gene × environment interaction? J Stud Alcohol Drugs. 2016;77(5):684–687. doi: 10.15288/jsad.2016.77.684
- Dick DM, Viken R, Purcell S, et al. Parental monitoring moderates the importance of genetic and environmental influences on adolescent smoking. J Abnorm Psychol. 2007;116(1):213–218. doi: 10.1037/0021-843X.116.1.213
- García-González J, Brock AJ, Parker MO, et al. Identification of slit3 as a locus affecting nicotine preference in zebrafish and human smoking behaviour. Elife. 2020;9:e51295. doi: 10.7554/eLife.51295
- Wang J, Yu XF, Ouyang N, et al. [Expression and prognosis effect of methylation-regulated SLIT3 and SPARCL1 genes in smoking-related lung adenocarcinoma]. Zhonghua Yi Xue Za Zhi. 2019;99(20):1553–1557. doi: 10.3760/cma.j.issn.0376-2491.2019.20.007
- Whang YM, Jo U, Sung JS, et al. Wnt5a is associated with cigarette smoke-related lung carcinogenesis via protein kinase C. PLoS One. 2013;8(1):e53012. doi: 10.1371/journal.pone.0053012
- Feller D, Kun J, Ruzsics I, et al. Cigarette smoke-induced pulmonary inflammation becomes systemic by circulating extracellular vesicles containing Wnt5a and inflammatory cytokines. Front Immunol. 2018;9:1724. doi: 10.3389/fimmu.2018.01724
- Hussain M, Rao M, Humphries AE, et al. Tobacco smoke induces polycomb-mediated repression of dickkopf-1 in lung cancer cells. Cancer Res. 2009;69(8):3570–3578. doi: 10.1158/0008-5472.CAN-08-2807
- Shen Z, He C, Chen H, et al. Potential genes associated with the survival of lung adenocarcinoma were identified by methylation. Comput Math Methods Med. 2020;2020:1–13. doi: 10.1155/2020/7103412
- Wain LV, Shrine N, Miller S, et al. Novel insights into the genetics of smoking behaviour, lung function, and chronic obstructive pulmonary disease (UK BiLEVE): a genetic association study in UK Biobank. Lancet Respir Med. 2015;3(10):769–781. doi: 10.1016/S2213-2600(15)00283-0
- Dijkstra AE, Postma DS, van Ginneken B, et al. Novel genes for airway wall thickness identified with combined genome-wide association and expression analyses. Am J Respir Crit Care Med. 2015;191(5):547–556. doi: 10.1164/rccm.201405-0840OC
- Quach BC, Bray MJ, Gaddis NC, et al. Expanding the genetic architecture of nicotine dependence and its shared genetics with multiple traits. Nat Commun. 2020;11(1):5562. doi: 10.1038/s41467-020-19265-z
- Koopmann A, Bez J, Lemenager T, et al. The effect of nicotine on HPA axis activity in females is modulated by the FKBP5 genotype. Ann Hum Genet. 2016;80(3):154–161. doi: 10.1111/ahg.12153
- Yeh JC, Hiraoka N, Petryniak B, et al. Novel sulfated lymphocyte homing receptors and their control by a Core1 extension β1,3-N-Acetylglucosaminyltransferase. Cell. 2001;105(7):957–969. doi: 10.1016/S0092-8674(01)00394-4
- Hennet T, Dinter A, Kuhnert P, et al. Genomic cloning and expression of three murine UDP-galactose: β-N-Acetylglucosamine β1,3-galactosyltransferase genes *. J Biol Chem. 1998;273(1):58–65. doi: 10.1074/jbc.273.1.58
- Leng X, Wei S, Mei J, et al. Identifying the prognostic significance of B3GNT3 with PD-L1 expression in lung adenocarcinoma. Transl Lung Cancer Res. 2021;10(2):965–980. doi: 10.21037/tlcr-21-146
- Gupta R, Leon F, Thompson CM, et al. Global analysis of human glycosyltransferases reveals novel targets for pancreatic cancer pathogenesis. Br J Cancer. 2020;122(11):1661–1672. doi: 10.1038/s41416-020-0772-3
- Zhuang H, Zhou Z, Zhang Z, et al. B3GNT3 overexpression promotes tumor progression and inhibits infiltration of CD8+ T cells in pancreatic cancer. Aging (Albany NY). 2021;13(2):2310–2329. doi: 10.18632/aging.202255
- Xu J, Guo Z, Yuan S, et al. Upregulation of B3GNT3 is associated with immune infiltration and activation of NF-κB pathway in gynecologic cancers. J Reprod Immunol. 2022;152:103658. doi:10.1016/j.jri.2022.103658
- Lu J, Lei T, Yu H, et al. The role of B3GNT3 as an oncogene in the growth, invasion and migration of esophageal cancer cells. Acta Cir Bras. 2023;38:e380923. doi: 10.1590/acb380923
- Zhou H, Zhao J, Yang X, et al. Study on the expression of β-1,3-N-acetylglucosaminyltransferase 3 in gastric cancer and the mechanism promoting gastric cancer progression based on the extraction method of Nanomagnetic Beads. J Biomed Nanotechnol. 2022;18(3):677–692. doi: 10.1166/jbn.2022.3296
- West KA, Brognard J, Clark AS, et al. Rapid akt activation by nicotine and a tobacco carcinogen modulates the phenotype of normal human airway epithelial cells. J Clin Invest. 2003;111(1):81–90. doi: 10.1172/JCI200316147
- Schuller HM. Mechanisms of smoking-related lung and pancreatic adenocarcinoma development. Nat Rev Cancer. 2002;2(6):455–463. doi: 10.1038/nrc824
- Philibert RA, Beach SRH, Lei M-K, et al. Changes in DNA methylation at the aryl hydrocarbon receptor repressor may be a new biomarker for smoking. Clin Epigenetics. 2013;5(1):19. doi: 10.1186/1868-7083-5-19
- Ambatipudi S, Cuenin C, Hernandez-Vargas H, et al. Tobacco smoking-associated genome-wide DNA methylation changes in the EPIC study. Epigenomics. 2016;8(5):599–618. doi: 10.2217/epi-2016-0001
- Qi S, Fu Z, Wu L, et al. Cognition, Aryl hydrocarbon receptor repressor methylation, and abstinence duration-associated multimodal brain networks in smoking and long-term smoking cessation. Front Neurosci. 2022;16:923065. doi: 10.3389/fnins.2022.923065
- Reynolds LM, Wan M, Ding J,et al. DNA methylation of the aryl hydrocarbon receptor repressor associations with cigarette smoking and subclinical atherosclerosis. Circulation. 2015;8:707–716. doi: 10.1161/CIRCGENETICS.115.001243
- Joubert BR, Håberg SE, Nilsen RM, et al. 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ Health Perspect. 2012;120(10):1425–1431. doi: 10.1289/ehp.1205412
- Lee KWK, Richmond R, Hu P, et al. Prenatal exposure to maternal cigarette smoking and DNA methylation: epigenome-wide association in a discovery sample of adolescents and replication in an Independent cohort at birth through 17 years of age. Environ Health Perspect. 2015;123(2):193–199. doi: 10.1289/ehp.1408614
- Richmond RC, Simpkin AJ, Woodward G, et al. Prenatal exposure to maternal smoking and offspring DNA methylation across the lifecourse: findings from the avon longitudinal study of parents and children (ALSPAC). Hum Mol Genet. 2015;24(8):2201–2217. doi: 10.1093/hmg/ddu739
- Ma Q. Induction of CYP1A1. The AhR/DRE Paradigm transcription, receptor regulation, and expanding biological roles. Curr Drug Metab. 2001;2(2):149–164. doi: 10.2174/1389200013338603
- Nebert DW, Roe AL, Dieter MZ, et al. Role of the aromatic hydrocarbon receptor and [ah] gene battery in the oxidative stress response, cell cycle control, and apoptosis. Biochem Pharmacol. 2000;59(1):65–85. doi: 10.1016/S0006-2952(99)00310-X
- Ohashi H, Nishioka K, Nakajima S, et al. The aryl hydrocarbon receptor–cytochrome P450 1A1 pathway controls lipid accumulation and enhances the permissiveness for hepatitis C virus assembly. J Biol Chem. 2018;293(51):19559–19571. doi: 10.1074/jbc.RA118.005033
- Tekpli X, Zienolddiny S, Skaug V, et al. DNA methylation of the CYP1A1 enhancer is associated with smoking-induced genetic alterations in human lung. Int J Cancer. 2012;131(7):1509–1516. doi: 10.1002/ijc.27421
- Tsai P-C, Glastonbury CA, Eliot MN, et al. Smoking induces coordinated DNA methylation and gene expression changes in adipose tissue with consequences for metabolic health. Clin Epigenet. 2018;10(1):126. doi: 10.1186/s13148-018-0558-0
- Mitsui Y, Chang I, Kato T, et al. Functional role and tobacco smoking effects on methylation of CYP1A1 gene in prostate cancer. Oncotarget. 2016;7(31):49107. doi: 10.18632/oncotarget.9470
- Thomas ET, Guppy M, Straus SE, et al. Rate of normal lung function decline in ageing adults: a systematic review of prospective cohort studies. BMJ Open. 2019;9(6):e028150. doi: 10.1136/bmjopen-2018-028150
- Schneider JL, Rowe JH, Garcia-de-Alba C, et al. The aging lung: physiology, disease, and immunity. Cell. 2021;184(8):1990–2019. doi: 10.1016/j.cell.2021.03.005
- Sharma G, Goodwin J. Effect of aging on respiratory system physiology and immunology. Clin Interv Aging. 2006;1(3):253–260. doi: 10.2147/ciia.2006.1.3.253
- Scanlon PD, Connett J, Waller L, et al. Smoking cessation and lung function in mild-to-moderate chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2000;161(2):381–390. doi: 10.1164/ajrccm.161.2.9901044
- Burchfiel CM, Marcus EB, Curb JD, et al. Effects of smoking and smoking cessation on longitudinal decline in pulmonary function. Am J Respir Crit Care Med. 1995;151(6):1778–1785. doi: 10.1164/ajrccm.151.6.7767520
- Bossé R, Sparrow D, Rose CL, et al. Longitudinal effect of age and smoking cessation on pulmonary function. Am Rev Respir Dis. 1981;123(4 Pt 1):378–381. doi: 10.1164/arrd.1981.123.4.378
- Oelsner EC, Balte PP, Bhatt SP, et al. Lung function decline in former smokers and low-intensity current smokers: a secondary data analysis of the NHLBI pooled cohorts study. Lancet Respir Med. 2020;8(1):34–44. doi: 10.1016/S2213-2600(19)30276-0
- Donaldson GC, Seemungal TAR, Patel IS, et al. Airway and systemic inflammation and decline in lung function in patients with COPD. Chest. 2005;128(4):1995–2004. doi: 10.1378/chest.128.4.1995
- Donaldson GC. Relationship between exacerbation frequency and lung function decline in chronic obstructive pulmonary disease. Thorax. 2002;57(10):847–852. doi: 10.1136/thorax.57.10.847
- Soria J-C, Rodriguez M, Liu DD, et al. Aberrant promoter methylation of multiple genes in bronchial brush samples from former cigarette smokers. Cancer Res. 2002;62(2):351–355.
- Belinsky SA, Palmisano WA, Gilliland FD, et al. Aberrant promoter methylation in bronchial epithelium and sputum from current and former smokers. Cancer Res. 2002;62(8):2370–2377.
- Wang G, Wang R, Strulovici-Barel Y, et al. Persistence of smoking-induced dysregulation of MiRNA expression in the small airway epithelium despite smoking cessation. PLoS One. 2015;10(4):e0120824. doi: 10.1371/journal.pone.0120824
- Hogg JC. Why does airway inflammation persist after the smoking stops? Thorax. 2006;61(2):96. doi: 10.1136/thx.2005.049502
- Rutgers S, Postma DS, Ten Hacken NH, et al. Ongoing airway inflammation in patients with COPD who do not currently smoke. Thorax. 2000;55(1):12. doi: 10.1136/thorax.55.1.12
- Shiels MS, Katki HA, Freedman ND, et al. Cigarette smoking and variations in systemic immune and inflammation markers. JNCI. 2014;106(11). doi: 10.1093/jnci/dju294
- Lapperre TS, Postma DS, Gosman MM, et al. Relation between duration of smoking cessation and bronchial inflammation in COPD. Thorax. 2006;61(2):115. doi: 10.1136/thx.2005.040519