
ABSTRACT
Drug resistance is the primary contributor to the high mortality rate of ovarian cancer (OC). The loss of BRCA1/2 function is linked to drug sensitivity in OC cells. The aim of this study is to enhance the drug sensitivity of OC cells by inducing BRCA1 dysfunction through promoter epigenetic editing. Epigenetic regulatory regions within the BRCA1 promoter, affecting gene expression, were initially discerned through analysis of clinical samples. Subsequently, we designed and rigorously validated epigenetic editing tools. Ultimately, we evaluated the cisplatin and olaparib sensitivity of the OC cells after editing. The BRCA1 promoter contains two CpG-rich regions, with methylation of the region covering the transcription start site (TSS) strongly correlating with transcription and influencing OC development, prognosis, and homologous recombination (HR) defects. Targeting this region in OC cells using our designed epigenetic editing tools led to substantial and persistent DNA methylation changes, accompanied by significant reductions in H3K27ac histone modifications. This resulted in a notable suppression of BRCA1 expression and a decrease in HR repair capacity. Consequently, edited OC cells exhibited heightened sensitivity to cisplatin and olaparib, leading to increased apoptosis rates. Epigenetic inactivation of the BRCA1 promoter can enhance cisplatin and olaparib sensitivity of OC cells through a reduction in HR repair capacity, indicating the potential utility of epigenetic editing technology in sensitization therapy for OC.
Introduction
The annual increase in ovarian cancer-related fatalities presents significant challenges in prevention and treatment. In 2022, ovarian cancer (OC) ranked as the fifth leading cause of cancer-related death among women in the United States [Citation1]. In China, in 2015, there were approximately 52,100 new cases of OC along with 22,500 fatalities, making it the seventh leading cause of cancer-related death among Chinese women [Citation2]. The most common histological subtype is high-grade serous ovarian cancer (HGSOC), initially responsive to platinum-based therapy [Citation3]. However, up to 75% of patients experience disease recurrence after remission, leading to platinum resistance. The survival rates for HGSOC have exhibited minimal alteration over the span of several decades [Citation3]. The 5-year relative survival rates for patients diagnosed between 2007 and 2013 were 42% and 26%, respectively [Citation4]. Progress in OC treatment has been limited, emphasizing the urgency of addressing drug resistance through innovative sensitization and pharmaceutical approaches.
Platinum-based chemotherapy is the gold standard for first-line treatment of OC [Citation5]. Drug resistance that occurs during chemotherapy may be due to genetic and epigenetic changes, assisting cancer cells in adapting to chemotherapy through various pathways such as stress, DNA damage, and apoptosis [Citation6]. Platinum-based chemotherapeutics trigger DNA interstrand cross-linking (ICL), inhibit transcription and replication, and promote cell cycle arrest and cell death through apoptosis. Activation of the DNA damage response (DDR) is crucial for tumour cell survival and the eventual development of platinum resistance [Citation7]. Platinum-induced ICLs can further progress to DNA double-strand breaks (DSBs), necessitating coordinated engagement of pathways like nucleotide excision repair (NER) and homologous recombination (HR) repair. While NER is essential for the removal of ICLs in quiescent cells, repair of ICLs in replicating cells relies on the HR pathway [Citation8]. Therefore, HR repair in tumours is closely associated with treatment outcomes. Currently, it is known that genetic mutations in 22 genes can lead to HR deficiency (HRD), including BRCA1/2, ATM, and CDK12 [Citation9]. The most common causes of HRD are germline BRCA mutations or somatic BRCA mutations [Citation10]. Moreover, abnormal epigenetic processes, such as DNA methylation and histone modifications, also play an important role in the initiation and progression of platinum resistance in OC [Citation11,Citation12]. DNA methyltransferases (DNMTs) are reported to be highly expressed in OC and are speculated to be associated with platinum resistance. Hypomethylating agents (HMAs) can resensitize ovarian tumours to platinum-based drugs and enhance clinical outcomes [Citation13–16]. Changes in DNMTs expression can lead to widespread alterations in the epigenome, including transcriptional inactivation of tumour suppressor genes (TSGs) [Citation14]. Numerous TSGs in OC exhibit high methylation and epigenetic silencing, including the hypermethylation of the BRCA1 promoter in 10–15% of OC tissues, which is associated with drug sensitivity [Citation16,Citation17].
Site-specific gene editing technology presents a promising avenue for ovarian cancer (OC) treatment, offering the potential to combat drug resistance and enhance patient outcomes [Citation18]. These innovative approaches encompass gene-specific knockouts and the regulation of gene expression [Citation18]. Notably, several studies have shown the potential of these approaches: CRISPR-Cas9-mediated knockdown of ABCB1 reversed doxorubicin resistance in OC cells [Citation19], BRCA1 knockout synergistically suppressed poly(ADP-ribose) polymerase (PARP)-1 expression in a mouse OC cell line (ID8), increasing sensitivity to cisplatin and PARP inhibitors [Citation20,Citation21]. Moreover, the stabilization of Snail, knockout of USP1, or the reduction of USP1 expression enhanced the sensitivity of tumour cells to chemotherapeutic agents [Citation22]. However, the lack of safe and effective delivery systems and off-target effects limit the further application of gene editing technologies [Citation23]. CRISPR-dCas9-based epigenetic editing may serve as an alternative approach to apply to sensitize chemotherapy for OC [Citation18]. Epigenetic editing offers a safer method for OC treatment, as it possesses three advantages not typically found in traditional gene editing: 1) independence from DNA repair pathways, high efficiency, and minimal side effects; 2) reversibility, in contrast to the essentially irreversible nature of gene editing; and 3) risk of off-target effects compared to traditional gene editing [Citation24]. Presently, epigenetic editing for chemotherapy sensitization encounters several challenges, encompassing a lack of adequate epigenetic editing tools, difficulties of in vivo targeted delivery, and the need to explore chemo-sensitivity epigenetic regulatory sites [Citation24].
In this investigation, we identified promoter regions responsible for epigenetically regulating BRCA1 expression and devised corresponding editing methods using the epigenetic editor CRISPRoff [Citation24]. After validating the editing effects, we found that epigenetic inactivation of BRCA1 in OC cells significantly enhanced their sensitivity to platinum and olaparib drugs
Materials and methods
DNA methylation analysis of OC samples in the dataset
The analysis of DNA methylation within the BRCA1 promoter and gene expression data in OC tumour samples utilized publicly available datasets from The Cancer Genome Atlas (TCGA). Correlation analysis between BRCA1 promoter methylation and gene expression in 398 TCGA Ovarian Serous Cystadenocarcinoma samples was conducted via cBioPortal (https://www.cbioportal.org/). The promoter region was defined as the region spanning 2000 bp upstream to 1000 bp downstream of the transcription start site (TSS). Data encompassing DNA methylation, SNP Array information, and survival data from 602 OC samples were obtained from TCGA (https://portal.gdc.cancer.gov). Survival rates associated with DNA methylation were estimated using the Kaplan-Meier (K-M) method, employing the survminer v0.4.9 R package. This package utilized the log-rank test for comparing survival times, with significance set at p < 0.05.
Plasmids construction
The Plasmid CRISPRoffv2.1 was acquired from Addgene (Addgene #167981). The sgRNA plasmids were generated by employing restriction cloning of protospacers downstream of a U6 promoter using the BsmBI cut site within the pLentiGuide-puro plasmid. It is noteworthy that the CRISPRoff plasmid also expresses a blue fluorescent protein (BFP) marker to facilitate the measurement of transfection efficiency. The selection of sgRNA sequences was guided by our previously established algorithm designed to predict active CRISPRi sgRNAs [Citation25]. The sgRNA sequences targeted the BRCA1 promoter were presented as follows: 5’-TGAAGGCCTCCTGAGCGCAG-3’ targeting chromosome 17 coordinates at position 41,277,324 and 5’-TTACCCAGAGCAGAGGGTGA-3’ targeting chromosome 17 coordinates at position 41,277,307.
Cell culture and transfections
Four human cell lines, 293T, A2780, OVCAR3, and SKOV3, were cultivated. The 293T cells were grown in Dulbecco’s Modified Eagle Medium (DMEM), while the OC cell lines A2780, OVCAR3, and SKOV3 were cultured in RPMI-1640 medium. These cell cultures were supplemented with 10% foetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S) antibiotics. The incubation conditions involved maintaining the cells at 37°C in an environment with 5% CO2 to ensure proper humidification. Transfections experiments in 293T, A2780, OVCAR3, and SKOV3 were carried out when the cells reached 70%–80% confluence, using 300 ng of CRISPRoff plasmid and 150 ng of sgRNA-encoding plasmids combined with Lipofectamine 3000 (Thermo Fisher Scientific, USA) in Opti-MEM medium in 24-well plates.
Clinical sample collection
For validation analysis, a collection of 81 OC formalin-fixed paraffin-embedded (FFPE) samples were obtained from the Xiangya Hospital, School of Basic Medical Sciences, Central South University (Changsha, China) between January 2017 and December 2020. The patients had an average age at diagnosis of 50.6 ± 11.0 years. Among these, we obtained 46 high-grade serous ovarian carcinoma (HGSOC) samples, along with matched peripheral blood samples for HRD analysis. Ethical approval for our study was granted by the local committee, and all participants provided written informed consent.
Calculation of HRD score and BRCA1/2 mutation analysis
Genomic scar signatures were employed to calculate the HRD score using an R program, as previously described [Citation26]. This calculation encompassed determining the Telomeric Allelic Imbalance (TAI), Large-Scale Transition (LST), and Loss of Heterozygosity (LOH) scores. TAI represented regions of allelic imbalance extending to the telomeres, LOH denoted chromosomal LOH regions longer than 15 Mb, and LST referred to breakpoints between regions longer than 10 Mb, after filtering out regions shorter than 3 Mb. The HRD score was computed as the sum of the TAI, LST, and LOH scores. In the TCGA cohort, HRD scores were calculated from SNP microarray data (Affymetrix GenomeWideSNP6 array). Both tumour and corresponding normal tissue was analysed. The same analytical process used for next-generation sequencing (NGS) was applied to analyse clinical tumour samples. The 3DMed Onco GPS Tissue NGS kit (3DMed, China) targeting 733 tumour-associated genes, including BRCA1/2, was used. Target genes were captured using probes and then sequenced on the HiSeq platform (Illumina, Inc., USA). BRCA1/2 germline mutations were also assessed using this gene panel. Briefly, raw sequencing data were aligned to the reference human genome (Feb. 2009, GRCh37/hg19) using BWA software (version 0.5.9) and SAMtools (version 0.1.18). Genome Analysis Toolkit (version 3.5) was used for variant calling, employing parameters such as UnifiedGenotyper, HaplotypeCaller, and VariantFiltration. Subsequently, ANNOVAR was applied for variant annotation. The interpretation of variants adhered to the American College of Medical Genetics and Genomics (ACMG) guidelines. Pathogenic or likely pathogenic variants encompassed frameshift insertions or deletions (indels), nonsense mutations, uncorrected splice-site variants, large-scale deletions, and missense mutations with impaired protein function, as determined by functional assays. Only pathogenic or likely pathogenic variants were classified as BRCA1/2 mutations.
Bisulfite amplicon sequencing (BSAS)
DNA from cell line samples and FFPE samples was extracted using the High Pure PCR Template Preparation Kit (Roche, USA) and GeneRead DNA FFPE Kit (QIAGEN, Germany), respectively. DNA methylation levels of candidate genes were assessed via bisulphite amplification and next-generation sequencing (NGS). Specifically, 10 ng of genomic DNA underwent bisulphite conversion with the EZ DNA Methylation-Lightning Kit (Zymo Research, USA). Subsequently, we performed PCR amplification of the target sequences separately and constructed the second-generation sequencing libraries with the TaKaRa EX Taq Hot Start Version Kit (TaKaRa, China). The primers employed for amplification were as follows: BRCA1 forward: 5’-GGAAAGGGATAGGGGGTTTAAGTGATGTT-3,’BRCA1 reverse: 5’-AATTCACAARGCCTTA-RGCCTCTCAAATTC-3.’ The library was 2 × 150 bp paired-end sequenced using the NGS HiSeq platform (Illumina, Inc., USA). The quality of the sequencing data was verified using the FastQC package. Adapter removal and quality trimming were performed using Cutadapt v1.9.1, the reads were aligned to a reference genome (UCSC hg19) using Bismark. Methylation levels of individual cytosines were determined through the utilization of the R package MethylKit v1.26.0.
Quantitative real time polymerase chain reactions
Total RNA was extracted from cultured cells and tissue samples using TRIzol Reagent (SIGMA, USA) according to the manufacturer’s instructions. Reverse transcription was performed with a mix of oligo dT primer and random primer for mRNA and with stem-loop primer for miRNA using the PrimeScriptRT Reagent Kit (TaKaRa, China). qPCR was performed with a standard three-step amplification protocol of SYBR Premix Ex TaqTM (TaKaRa, China). qPCR reactions were performed by using CFX96 Real-Time PCR detector (Bio-Rad, USA) and SYBR Green PCR Mix (TaKaRa, China). And the comparative Cq method was used to calculate the fold change. Amplification conditions were 60 min at 60°C, then 5 min at 95°C, 15 s at 95°C, and 1 min at 60°C, 45 PCR cycles. BRCA1 primer (forward, 5’-ACAGCTGTGTGGTGCTTCTGTG-3,“ reverse 5”-CATTGTCCTCTGTCCA-GGCATC-3“), GAPDH primer (forward 5”-TCATTGACCTCAACTACATGGTTT-3,“ reverse 5”- GAAGATGGTGATGGGGATTTC-3’).
Chromatin immunoprecipitation (ChIP)-qPCR
A2780 cells were briefly chilled in 125 mM glycine at room temperature for 5 minutes following fixation in 1% formaldehyde for 10 minutes. After fixation, the cells were subjected to centrifugation at 1,000 g for 10 minutes at 4°C and washed thrice with PBS. Subsequently, cell pellets were resuspended in 10 mL of ChIP lysis buffer (comprising 10 mM Tris-HCl at pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Igepal, and protease inhibitors) and were incubated at 4°C for 60 minutes on a rotary shaker. The nuclei were then isolated by centrifugation (10 minutes, 4°C, 1,000 g), reconstituted in SDS lysis buffer (containing 1% SDS, 10 mM EDTA, and 50 mM Tris-HCl at pH 8.0), and incubated on ice for 30 minutes. SDS was diluted by adding 1× TE, and sonication was performed using a Covaris M220 (Covaris, Inc., USA). The efficiency of sonication was assessed via agarose gel electrophoresis. Subsequently, ChIP assays were conducted at 4°C for 16 hours using 2 μg of H3K27ac antibody (A7253, ABclonal, China). Immune complexes were captured with 20 μl of magnetic protein A/G beads (Thermo Fisher Scientific, USA) for 2 hours at 4°C. The beads underwent two washes with RIPA, three washes with ChIP wash buffer (containing 100 mM Tris pH 8, 500 mM LiCl, and 1% deoxycholic acid), and one wash with ChIP wash buffer supplemented with 150 mM NaCl. ChIP complexes were eluted in 100 μl of ChIP elution buffer (comprising 50 mM NaHCO3 and 1% SDS) for 30 minutes, and cross-links were reversed in the presence of 0.5 M NaCl overnight at 65°C. ChIP DNA was purified using the QIAquick PCR Purification Kit (QIAGEN, Germany). Quantitative PCR was performed on ChIP and input control samples using the SYBR Premix Ex TaqTM (TaKaRa, China) and the CFX96 Real-Time PCR detector (Bio-Rad, USA) with SYBR Green PCR Mix (TaKaRa, China). The ChIP amplification primers targeting the BRCA1 promoter are as follows: 5’-CGTATTCTGAGAGGCTGCTG-3’ and 5’-GGAAGTCTCAGCGAGCTCAC-3.’ ChIP enrichment was determined relative to input samples using the dCq method (dCq = Cq[ChIP] - Cq[input]).
Cell proliferation assay
Twenty-four hours prior to transfection, A2780 cells were seeded in 6-well plates and allowed to reach a confluence of 70%-90%. Transfection was performed using Lipofectamine 3000 (Thermo Fisher Scientific, USA) with a co-transfection of methylation editing plasmid pLentiGuide-puro-BRCA1 and CRISPRoff-v2.1 (500 ng each per well). Plasmids without editing effects were used as controls. Twenty-four hours after transfection, cells were collected and seeded at a density of 2 × 104 cells per well in a 96-well plate. After 24 hours of plating, different concentrations of cisplatin (DDP) (0.75 μg/ml and 1 μg/ml)(SIGMA, USA) and olaparib (10 μM and 20 μM) (MedChemExpress, USA) were added for drug sensitivity experiments. Cell Counting Kit-8 (DOJINDO, China) was added at days 0, 1, 3, 4, and 7 post-drug administration. Optical density at 450 nm (OD450) was measured after a 2-hour incubation period to analyse cell proliferation.
Detection of cell apoptosis
After trypsin digestion of adherent cells without EDTA, cells were collected by centrifugation at 300 × g for 5 minutes at 4°C. Apoptosis rates were measured by Cell Cycle and Apoptosis Analysis Kit (Yeasen, China). Specifically, cells were washed twice with pre-cooled PBS. Approximately 1–5 × 105 cells were collected. PBS was aspirated, and cells were resuspended in 100 μL of 1× Binding Buffer. Five microlitres of Annexin V-FITC and 10 μL of PI Staining Solution were added and gently mixed. The mixture was incubated in the dark at room temperature for 10–15 minutes. After adding 400 μL of 1× Binding Buffer and thorough mixing, the sample was placed on ice and analysed using a flow cytometer within 1 hour.
Homologous recombination (HR) assay
To assess HR capacity in A2780 and SKOV3 cells, a dual-immunofluorescence staining of γH2AX and RAD51 was conducted. Cells were transfected with pLentiGuide-puro-BRCA1 and CRISPRoffv2.1 or control plasmids. Following a 5-day incubation, cells were exposed to 10 μM DDP (SIGMA, USA) for 24 hours. Subsequently, the cells were subjected to nuclear extraction with buffer containing 20 mM HEPES (pH 7.4), 20 mM NaCl, 5 mM MgCl2, 0.5% NP-40, 1 mM DTT, and protease inhibitors for 10 minutes on ice. Afterwards, they were rinsed with ice-cold PBS and fixed with 4% PFA for 10 minutes at room temperature. The cells were then blocked in an immunofluorescence (IF) blocking buffer composed of 10% goat serum, 0.5% NP-40, and 0.5% saponin in PBS for 30 minutes. Subsequently, they were incubated with primary antibodies: mouse anti-RAD51 (GeneTex, USA) and rabbit anti-γ-H2AX (Cell Signaling, USA) antibodies diluted in blocking buffer for 2 hours at room temperature. Following primary antibody incubation, the cells were washed with PBS (3× 5 minutes) and stained with fluorescent secondary antibodies: Goat anti-Rabbit IgG (H+L) Secondary Antibody, DyLight 488 (Thermo Fisher Scientific, USA) and Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 (Thermo Fisher Scientific, USA) for 1 hour at room temperature. After the secondary antibody staining, cells were washed as previously described, mounted in ProLong Gold mounting media (Thermo Fisher Scientific, USA), and imaged using a Ti-E+A1R+STORM10–1 (Nikon, Japan). Data were collected from 100 cells in five to ten fields in two independent experiments.
Statistical analysis
Graphs and statistical tests were analysed using GraphPad Prism version 9.0 (GraphPad Software, USA). Non-parametric Mann-Whitney test or unpaired t-test was utilized to compare the mean values of the control and tested groups, and p < 0.05 was considered significant.
Results
Analysis of DNA methylation characteristics in the BRCA1 gene promoter in OC
BRCA1 is situated at 17q21.31, with a TSS positioned at 41,277,500 (Hg19) in the reverse orientation. Adhering to the promoter’s definition, we selected a DNA segment ranging from 2,000 bp upstream to 1,000 bp downstream of the TSS (41,276,500–41,279,500) for scrutiny. Within this promoter region, there are three CpG islands, located at 41,279,075–41,277,986, 41,277,694–41,277,244, and 41277,206–41,276,698. The latter two CpG islands are contiguous, forming a region encompassing the TSS, exon 1, and part of intron 1. This region, along with the first CpG island, constitutes two CG-dense regions, in which there are eight CG dinucleotides with methylation data in the TCGA OC database (). Analysis of methylation levels of these CGs and their correlation with gene expression in the TCGA OC datasets unveiled that six CGs proximal to the TSS were predominantly hypomethylated, with a minority exhibiting hypermethylation in tumour tissues. In contrast, DNA methylation levels of the two CGs within the CpG island, 1000 base pairs upstream of the TSS, were generally high, with a few samples displaying hypomethylation. Moreover, DNA methylation levels of six CGs in the region near the TSS exhibited a highly significant negative correlation with gene expression, while the correlation between methylation and expression of two CGs upstream of the TSS was negligible or weak ().
Figure 1. The relationship between methylation at CG sites within the BRCA1 promoter and gene expression levels.
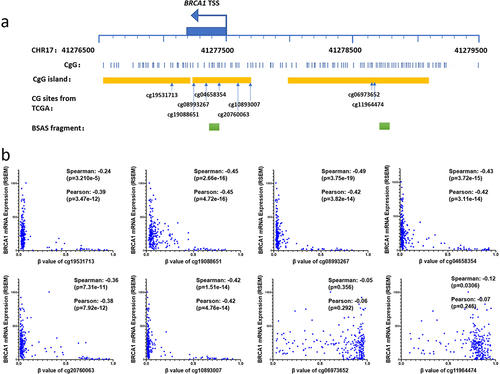
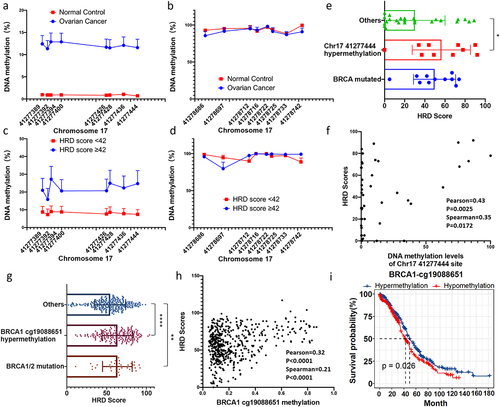
We collected OC tissue samples and conducted a comprehensive analysis of DNA methylation levels in the two regions of the BRCA1 promoter. Employing the BSAS assay, we amplified and sequenced two segments within these regions located on chromosome 17: 41277,787–41,277,627 and 41,275,000–41,277,199, respectively (). The analysis revealed that DNA methylation levels of all eight CGs proximate to the TSS were less than 1% in normal ovarian tissues. Conversely, within OC tissues, certain samples exhibited elevated methylation levels, with an average methylation level ranging from 10% to 15%, thereby signifying a noteworthy disparity between these two groups (). Conversely, the eight CGs within the CpG island upstream of the TSS were hypermethylated both in normal ovarian tissues and OC tissues, with no significant difference between the two groups (). These results are consistent with TCGA sample results. It can be inferred that the methylation levels of CGs near the TSS are intricately linked to the expression level of the tumour suppressor gene BRCA1, and escalated methylation levels may be associated with the inactivation of the BRCA1 promoter in OC. Furthermore, we examined BRCA1 promoter methylation in clinical cases, dividing them into HRD-positive (HRD score ≥ 42) and HRD-negative (HRD score < 42) groups. HRD-positive samples showed significantly higher DNA methylation levels at eight CG sites near the TSS compared to HRD-negative samples (). In contrast, methylation levels of eight CG sites within the CpG islands upstream of the TSS were similar across different HRD score groups (). Notably, both in our OC samples and the TCGA OC samples, samples with hypermethylation (>20%) at promoter CG sites displayed higher HRD scores, with mean values approaching or exceeding those of samples harbouring mutations in the BRCA1/2 genes (, g). Furthermore, a significant positive correlation between promoter CG site methylation levels and HRD scores was observed (, h). Survival data analysis of TCGA samples found that hypermethylation in the region near the TSS was associated with higher survival rate (). These results further suggest that DNA methylation levels in the region near the TSS are associated with homologous recombination defects and may affect survival time through differential response to treatment.
Figure 2. Methylation status of the BRCA1 promoter and its implications for OC development and homologous recombination defects (HRD).
Design and validation of the epigenetic editor for the BRCA1 promoter
The epigenetic editing plasmid CRISPRoff (DNMT3A-DNMT3L-XTEN80-dCas9-HA-2×NLS-BFP-KRAB) serves to augment DNA methylation while diminishing histone modifications like H3K27ac, with the ultimate objective of repressing gene expression. Our approach involved the design of two sgRNAs (targeting chromosome 17 coordinates at positions 41,277,324 and 41,277,307, respectively) with a specific focus on the BRCA1 TSS region, followed by the construction of the pLentiGuide-puro plasmids for precise site-specific epigenetic modification (). Co-transfection of CRISPRoff plasmid and pLentiGuide-puro plasmids was carried out in 293T, A2780, and OVCAR3 cells. Subsequently, we conducted a five-day observation of the fluorescent protein expression on the plasmids using fluorescence microscopy, aimed at assessing expression efficiency and stability. The results distinctly indicated the complete disappearance of fluorescent protein expression on the fifth day, signifying that the epigenetic editing plasmid exhibited transient expression within the cells.
Figure 3. Design and validation of the epigenetic editor for the BRCA1 promoter.
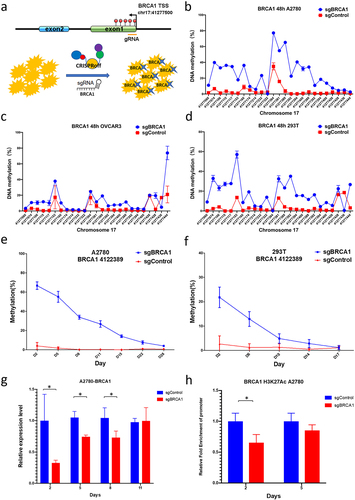
We further examined changes in DNA methylation levels after editing. A significant escalation in DNA methylation levels was observed within a 400bp range upstream and downstream of the sgRNA binding site at 48 hours post transfection in OC cells A2780 and OVCAR3 cells as well as in 293T cells (). The elevation in DNA methylation is higher in A2780 and 293T cells, some CGs had increase of methylation more than 50%. However, most of the CGs in OVCAR3 cells had less increased methylation levels, between 5–20%. Notably, minimal changes in DNA methylation occurred at the sgRNA binding site itself. Continuous cultivation of edited cells unveiled a gradual reduction in DNA methylation; however, it is worth noting that in A2780 cells, the DNA methylation level within the TSS region remained significantly elevated compared to the control group even on the 28th day (), whereas 293T cells sustained heightened methylation for a duration of 14 days (). In addition, analysis of BRCA1 gene expression in A2780 cells showed significantly lower mRNA levels than in unedited control cells from the second to the eighth day after editing (). Regarding the histone modification in A2780 cells, we found that the level of H3K27ac modification was significantly reduced compared to controls (). Collectively, these findings unequivocally substantiate our achievement of BRCA1 expression suppression in ovarian cancer cells via epigenetic editing.
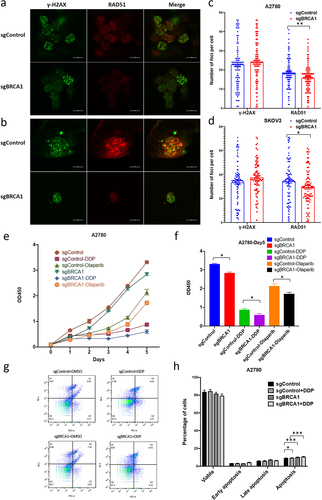
Epigenetic editing of the BRCA1 promoter affects HR repair capacity and increases drug sensitivity in OC cells
After confirming significant epigenetic modification changes and gene expression alterations in the BRCA1 promoter region after editing, we examined the altered HR repair capacity of the cells and their altered sensitivity to platinum-based drugs and poly(ADP-ribose) polymerase inhibitors (PARPi). We first determined the appropriate concentrations of cisplatin and olaparib in A2780 cells and found that 1 μg/ml of cisplatin and 10 μM olaparib inhibited approximately about half of the cell growth after five days by cell proliferation assay, which was selected as the subsequent experimental drug concentration (data not shown). Cells were transfected with the BRCA1 epigenetic editing plasmid for 5 days, 1 μg/ml of cisplatin was added, and HR capacity was determined by Dual-immunofluorescence staining of γH2AX/RAD51 after 1 day of treatment. Foci of γH2AX represent the location on the chromosome where the double-strand breaks have occurred, and the RAD51 foci represent ongoing HR repair. The ratio of the two co-localizations represents the HR repair capacity. The results showed that the RAD51 foci of BRCA1 promoter-edited cells treated with drugs were significantly reduced in both OC cells, A2780 () and SKOV3 (). Counting results showed that in A2780 cells, the mean number of RAD51 foci in edited A2780 cells was 15.8 compared to the mean number of 18.7 in unedited controls (), whereas in SKOV3 the mean number of foci in unedited and edited cells was 34.5 and 29.3, respectively (). These results suggest that epigenetic inactivation of the BRCA1 promoter would reduce HR repair capacity. For drug sensitivity test, A2780 cells were treated with 1 μg/ml of cisplatin and 10 μM olaparib for five consecutive days 48 hours after epigenetic editing, and cell counts showed a significant reduction in cell proliferation after epigenetic editing of the BRCA1 promoter (). The number of cells with promoter epigenetic editing was reduced by 32% on day five after cisplatin treatment compared to unedited cells, while it was reduced by 19% in the olaparib-treated group (). The above results indicate that the increased epigenetic modifications in the BRCA1 promoter region that inhibit expression lead to a significant increase in cellular sensitivity to platinum-based and PARPi drugs. Interestingly in the control group without drug treatment, the number of cells in the sgBRCA1 group was reduced by 14% on day 5, which was also significantly lower than in the control group (). We analysed the apoptosis of cells after drug treatment (), and the percentage of apoptosis was significantly higher in A2780 cells epigenetically edited for the BRCA1 promoter under DDP treatment than in unedited cells (). Even in the absence of drug treatment, epigenetically edited cells still exhibited a higher percentage of apoptosis (). This result suggested that epigenetic editing of the BRCA1 promoter may increase drug sensitivity by promoting apoptosis.
Figure 4. The impact of epigenetic editing of the BRCA1 promoter on HR repair capacity and drug sensitivity in OC cells.
Discussion
Treatment resistance in OC is a challenging issue that significantly impacts patient prognosis. While mechanisms behind this resistance have been widely studied, effective strategies to reverse or sensitize drug-resistant tumours remain elusive. This investigation seeked to leverage epigenetic editing techniques to induce hypermethylation in the BRCA1 promoter, thereby enhancing cellular drug sensitivity. Through the analysis of databases and our clinical samples, we identified critical promoter regions influenced by DNA methylation that profoundly affect BRCA1 transcription. We designed editing tools to increase DNA methylation in this region and remove H3K27ac modifications, resulting in sustained suppression of BRCA1 expression. Consequently, edited cells reduced HR repair capacity, promoted apoptosis, and had significantly increased sensitivity to platinum and PARPi drugs. These findings validate the relationship between epigenetic modifications of the BRCA1 promoter and platinum or PARPi resistance in OC, suggesting that targeted epigenetic regulation of BRCA1 represents a viable strategy for sensitizing OC to treatment.
In addition to mutations, the inactivation of the BRCA1 promoter leads to reduced BRCA1 expression, correlating with increased risks of ovarian and breast cancers. Numerous studies have indicated that promoter methylation of BRCA1 is a common occurrence in OC, particularly in HGSOC [Citation27,Citation28]. BRCA1 functional loss, including promoter methylation, leads to HRD [Citation29,Citation30]. Approximately 50% of HGSOC exhibit HRD, representing a distinct subtype with significant clinical implications, particularly in terms of treatment. HRD OCs show increased sensitivity to platinum-based chemotherapy, making this therapeutic approach preferable. Additionally, novel targeted drugs, such as PARPi, have demonstrated marked cytotoxic effects on HRD tumour cells [Citation31–34]. HR operates during the S and G2 phases of the cell cycle, relying on numerous proteins, including BRCA1 and BRCA2 [Citation30,Citation35]. When cells lack functional HR, often due to BRCA1 or BRCA2 deficiency, they rely on alternative repair pathways like non-homologous end-joining (NHEJ), which are less precise and prone to errors [Citation36].
Clinical studies have found that OC with hypermethylation in the BRCA1 promoter shares similar clinical and pathological features with OC harbouring BRCA1/2 gene mutations [Citation27]. Although reported occurrence rates range from 5% to 89.9% [Citation37,Citation38], it is estimated that approximately 10%-15% of OC cases exhibit BRCA1 promoter hypermethylation [Citation10,Citation39–41]. Additionally, BRCA1/2 germline mutations and somatic mutations can be observed in 15%-20% of OC cases [Citation10,Citation42]. BRCA1/2 mutations, along with BRCA1 promoter hypermethylation, are associated with significantly higher response rates and extended progression-free survival in OC patients following platinum-based chemotherapy [Citation10,Citation43,Citation44]. Analyses of BRCA1 methylation in OC samples consistently demonstrate lower levels of BRCA1 protein and mRNA expression [Citation40,Citation45–47]. Cell line models of BRCA1 methylation in OC exhibit specific sensitivity to platinum-based chemotherapy and PARP inhibitors [Citation48]. Xenograft models of OC derived from cell lines and patients with BRCA1 hypermethylation demonstrate significant sensitivity to platinum and/or PARP inhibitor therapy [Citation48–50]. These reports align with our findings.
The contribution of hypermethylation of promoter and mutations in BRCA1 to drug resistance differs. Several studies have shown that genotoxic drugs can lead to promoter demethylation, re-expression of BRCA1, and consequently, drug resistance. Exposure of BRCA1-highly methylated triple-negative breast cancer (TNBC) cells to platinum-based chemotherapy, whether as a clinical treatment or experimental in vivo exposure as patient-derived xenografts (PDX), results in promoter methylation loss, increased BRCA1 expression, and platinum resistance [Citation51]. Several studies have found that substances such as bisphenol A (BPA) and curcumin led to the re-expression of BRCA1 in cancer cell lines with hypermethylation in the BRCA1 promoter [Citation52,Citation53]. These results suggest that cancer cases with hypermethylation in the BRCA1 promoter exhibit a high adaptability to environmental exposure. By reversing promoter methylation level, BRCA1 expression can be restored, leading to therapy resistance [Citation51]. Therefore, in samples without BRCA mutations, the methylation status of this gene’s promoter is a key factor in drug sensitivity. Novel approaches targeting epigenetic modifications of the BRCA1 promoter may offer a new path to overcome resistance.
Epigenetic modifications are reversible and are carried out by various epigenetic modifying enzymes within cells. This makes it possible to regulate BRCA1 expression through epigenetic editing. Developments in epigenetic editing techniques have revealed that simultaneous modification of DNA methylation and histone tail amino acids can produce sustained modification effects and effectively regulate gene expression. In OC, over half of the cases lack BRCA mutations and do not exhibit hypermethylation in the BRCA promoter. Epigenetic editing to induce promoter inactivation may benefit this subset of patients in chemotherapy. Additionally, some patients who have BRCA1 reactivated during treatment may potentially benefit from re-closing BRCA1 expression through epigenetic editing of the promoter. Our experimental results provide preliminary evidence that this strategy is feasible.
Certainly, our strategy requires validation in animal models. Furthermore, the method of in vivo drug administration presents another challenge, with potential approaches including viral vectors and non-viral lipid nanoparticles, among others.
Conclusion
In summary, we employed epigenetic editing to deactivate the BRCA1 promoter, thereby enhancing the sensitivity of OC cells to cisplatin and olaparib. Initially, we identified crucial regions in the BRCA1 promoter that are epigenetically controlled, both in TCGA ovarian cancer samples and our own clinical cases. Subsequently, we devised targeted epigenetic editing tools. Following the confirmation of the effectiveness of these edits in OC cells, we assessed HR repair capacity, apoptosis rates, and drug sensitivity. The results showed that inactivation of the BRCA1 promoter through epigenetic editing can heighten cellular responsiveness to cisplatin and olaparib. It is imperative to undertake further in vivo experiments for validation.
Availability of data and materials
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Author contributions
XN and QW designed the study. WH, HZ and QW collected the data from databases and performed analyses. WH, HZ and QW organized and arranged all the figures. WH, HZ and SZ performed the experiments and the formal analysis. GS, HL, MW and GY collected tissue samples. WH and QW prepared and wrote the original draft of the manuscript. XN reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
Ethics approval and consent to participate
The studies involving human participants were reviewed and approved by the Xiangya School of Medicine, Central South University (approval no. 2019030306). The patients/participants provided their written informed consent to participate in this study. The study was conducted in accordance with the Declaration of Helsinki.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Siegel RL, Miller KD, Fuchs HE, et al. Cancer statistics, 2022. CA Cancer J Clin. 2022 Jan;72(1):7–15. doi: 10.3322/caac.21708
- Chen W, Zheng R, Baade PD, et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66(2):115–132. doi: 10.3322/caac.21338
- Vaughan S, Coward JI, Bast RC, et al. Rethinking ovarian cancer: recommendations for improving outcomes. Nat Rev Cancer. [2011 Sep 23];11(10):719–725. doi: 10.1038/nrc3144
- Torre LA, Trabert B, DeSantis CE, et al. Ovarian cancer statistics, 2018. CA Cancer J Clin. 2018 Jul;68(4):284–296. doi: 10.3322/caac.21456
- Lheureux S, Braunstein M, Oza AM. Epithelial ovarian cancer: Evolution of management in the era of precision medicine. CA Cancer J Clin. 2019 Jul;69(4):280–304. doi: 10.3322/caac.21559
- Pokhriyal R, Hariprasad R, Kumar L, et al. Chemotherapy resistance in advanced ovarian cancer patients. Biomark Cancer. 2019;11:1179299X19860815. doi: 10.1177/1179299X19860815
- Aguilera A, Gómez-González B. Genome instability: a mechanistic view of its causes and consequences. Nat Rev Genet. 2008 Mar;9(3):204–217. doi: 10.1038/nrg2268
- Reed E. Platinum-DNA adduct, nucleotide excision repair and platinum based anti-cancer chemotherapy. Cancer Treat Rev. 1998 Oct;24(5):331–344. doi: 10.1016/s0305-7372(98)90056-1
- Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer. 2016 Feb;16(2):110–120. doi: 10.1038/nrc.2015.21
- Network CGAR. Integrated genomic analyses of ovarian carcinoma. Nature. [2011 Jun 29];474(7353):609–615. doi: 10.1038/nature10166
- Oza AM, Matulonis UA, Alvarez Secord A, et al. A randomized phase II trial of epigenetic priming with guadecitabine and carboplatin in platinum-resistant, recurrent ovarian cancer. Clin Cancer Res. [2020 Mar 1];26(5):1009–1016. doi: 10.1158/1078-0432.CCR-19-1638
- Cardenas H, Fang F, Jiang G, et al. Methylomic signatures of high grade serous ovarian cancer. Epigenetics. 2021 Nov;16(11):1201–1216. doi: 10.1080/15592294.2020.1853402
- Cao YL, Zhuang T, Xing BH, et al. Exosomal DNMT1 mediates cisplatin resistance in ovarian cancer. Cell Biochem Funct. 2017 Aug;35(6):296–303. doi: 10.1002/cbf.3276
- Balch C, Fang F, Matei DE, et al. Minireview: epigenetic changes in ovarian cancer. Endocrinology. 2009 Sep;150(9):4003–4011. doi: 10.1210/en.2009-0404
- Fiegl H, Windbichler G, Mueller-Holzner E, et al. HOXA11 DNA methylation–a novel prognostic biomarker in ovarian cancer. Int J Cancer. [2008 Aug 1];123(3):725–729. doi: 10.1002/ijc.23563
- Loveday C, Turnbull C, Ramsay E, et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat Genet. [2011 Aug 7];43(9):879–882. doi: 10.1038/ng.893
- Meindl A, Hellebrand H, Wiek C, et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat Genet. 2010 May;42(5):410–414. doi: 10.1038/ng.569
- Luo S, Wang Y, Tao Y, et al. Application in gene editing in ovarian cancer therapy. Cancer Invest. 2022 Apr;40(4):387–399. doi: 10.1080/07357907.2021.1998521
- Norouzi-Barough L, Sarookhani M, Salehi R, et al. CRISPR/Cas9, a new approach to successful knockdown of ABCB1/P-glycoprotein and reversal of chemosensitivity in human epithelial ovarian cancer cell line. Iran J Basic Med Sci. 2018 Feb;21(2):181–187. doi: 10.22038/IJBMS.2017.25145.6230
- Walton JB, Farquharson M, Mason S, et al. CRISPR/Cas9-derived models of ovarian high grade serous carcinoma targeting Brca1, Pten and Nf1, and correlation with platinum sensitivity. Sci Rep. [2017 Dec 4];7(1):16827. doi: 10.1038/s41598-017-17119-1
- Kim SM, Yang Y, Oh SJ, et al. Cancer-derived exosomes as a delivery platform of CRISPR/Cas9 confer cancer cell tropism-dependent targeting. J Control Release. [2017 Nov 28];266:8–16. doi: 10.1016/j.jconrel.2017.09.013
- Sonego M, Pellarin I, Costa A, et al. USP1 links platinum resistance to cancer cell dissemination by regulating snail stability. Sci Adv. 2019 May;5(5):eaav3235. doi: 10.1126/sciadv.aav3235
- Li L, He ZY, Wei XW, et al. Challenges in CRISPR/CAS9 delivery: Potential roles of nonviral vectors. Hum Gene Ther. 2015 Jul;26(7):452–462. doi: 10.1089/hum.2015.069
- Nuñez JK, Chen J, Pommier GC, et al. Genome-wide programmable transcriptional memory by CRISPR-based epigenome editing. Cell. [2021 Apr 29];184(9):2503–2519. e17 doi:10.1016/j.cell.2021.03.025
- Horlbeck MA, Gilbert LA, Villalta JE, et al. Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. Elife. [2016 Sep 23]:5. doi: 10.7554/eLife.19760
- Marquard AM, Eklund AC, Joshi T, et al. Pan-cancer analysis of genomic scar signatures associated with homologous recombination deficiency suggests novel indications for existing cancer drugs. Biomark Res. 2015;3(1):9. doi: 10.1186/s40364-015-0033-4
- Kalachand RD, Stordal B, Madden S, et al. BRCA1 promoter methylation and clinical outcomes in ovarian cancer: An individual patient data meta-analysis. J Natl Cancer Inst. [2020 Dec 14];112(12):1190–1203. doi: 10.1093/jnci/djaa070
- Kondrashova O, Topp M, Nesic K, et al. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat Commun. [2018 Sep 28];9(1):3970. doi: 10.1038/s41467-018-05564-z
- Ledermann JA, Drew Y, Kristeleit RS. Homologous recombination deficiency and ovarian cancer. Eur J Cancer. 2016 Jun;60:49–58. doi: 10.1016/j.ejca.2016.03.005
- Moschetta M, George A, Kaye SB, et al. BRCA somatic mutations and epigenetic BRCA modifications in serous ovarian cancer. Ann Oncol. 2016 Aug;27(8):1449–1455. doi: 10.1093/annonc/mdw142
- Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. [2005 Apr 14];434(7035):917–921. doi: 10.1038/nature03445
- Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. [2009 Jul 9];361(2):123–134. doi: 10.1056/NEJMoa0900212
- Jagtap P, Szabó C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat Rev Drug Discov. 2005 May;4(5):421–440. doi: 10.1038/nrd1718
- Ratnam K, Low JA. Current development of clinical inhibitors of poly(ADP-ribose) polymerase in oncology. Clin Cancer Res. [2007 Mar 1];13(5):1383–1388. doi: 10.1158/1078-0432.CCR-06-2260
- Lupo B, Trusolino L. Inhibition of poly(ADP-ribosyl)ation in cancer: old and new paradigms revisited. Biochim Biophys Acta. 2014 Aug;1846(1):201–215. doi: 10.1016/j.bbcan.2014.07.004
- Wang M, Wu W, Rosidi B, et al. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006;34(21):6170–6182. doi: 10.1093/nar/gkl840
- Catteau A, Harris WH, Xu CF, et al. Methylation of the BRCA1 promoter region in sporadic breast and ovarian cancer: correlation with disease characteristics. Oncogene. [1999 Mar 18];18(11):1957–1965. doi: 10.1038/sj.onc.1202509
- Pradjatmo H, Dasuki D, Anwar M, et al. Methylation status and immunohistochemistry of BRCA1 in epithelial ovarian cancer. Asian Pac J Cancer Prev. 2014;15(21):9479–9485. doi: 10.7314/apjcp.2014.15.21.9479
- Abkevich V, Timms KM, Hennessy BT, et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br J Cancer. [2012 Nov 6];107(10):1776–1782. doi: 10.1038/bjc.2012.451
- Baldwin RL, Nemeth E, Tran H, et al. BRCA1 promoter region hypermethylation in ovarian carcinoma: a population-based study. Cancer Res. [2000 Oct 1];60(19):5329–5333.
- Geisler JP. Frequency of BRCA1 dysfunction in ovarian cancer. J Natl Cancer Inst. [2002 Jan 2];94(1):61–67. doi: 10.1093/jnci/94.1.61
- Hennessy BT, Lu Y, Poradosu E, et al. Pharmacodynamic markers of perifosine efficacy. Clin Cancer Res. [2007 Dec 15];13(24):7421–7431. doi: 10.1158/1078-0432.CCR-07-0760
- Bolton KL, Chenevix-Trench G, Goh C, Sadetzki S, Ramus SJ, Karlan BY, Lambrechts D, Despierre E, Barrowdale D, McGuffog L, Healey S. Association between BRCA1 and BRCA2 mutations and survival in women with invasive epithelial ovarian cancer. JAMA. [2012 Jan 25];307(4):382–390. doi: 10.1001/jama.2012.20
- Yang D, Khan S, Sun Y, et al. Association of BRCA1 and BRCA2 mutations with survival, chemotherapy sensitivity, and gene mutator phenotype in patients with ovarian cancer. JAMA. [2011 Oct 12];306(14):1557–1565. doi: 10.1001/jama.2011.1456
- Press JZ, De Luca A, Boyd N, et al. Ovarian carcinomas with genetic and epigenetic BRCA1 loss have distinct molecular abnormalities. BMC Cancer. [2008 Jan 22];8:17. doi: 10.1186/1471-2407-8-17
- Swisher EM, Gonzalez RM, Taniguchi T, et al. Methylation and protein expression of DNA repair genes: association with chemotherapy exposure and survival in sporadic ovarian and peritoneal carcinomas. Mol Cancer. [2009 Jul 14];8:48. doi: 10.1186/1476-4598-8-48
- Wang C, Horiuchi A, Imai T, et al. Expression of BRCA1 protein in benign, borderline, and malignant epithelial ovarian neoplasms and its relationship to methylation and allelic loss of the BRCA1 gene. J Pathol. 2004 Feb;202(2):215–223. doi: 10.1002/path.1507
- Stordal B, Timms K, Farrelly A, et al. BRCA1/2 mutation analysis in 41 ovarian cell lines reveals only one functionally deleterious BRCA1 mutation. Mol Oncol. 2013 Jun;7(3):567–579. doi: 10.1016/j.molonc.2012.12.007
- Veeck J, Ropero S, Setien F, et al. BRCA1 CpG island hypermethylation predicts sensitivity to poly(adenosine diphosphate)-ribose polymerase inhibitors. J Clin Oncol. [2010 Oct 10];28(29):e563–4; author reply e565-6. doi:10.1200/JCO.2010.30.1010
- Topp MD, Hartley L, Cook M, et al. Molecular correlates of platinum response in human high-grade serous ovarian cancer patient-derived xenografts. Mol Oncol. 2014 May;8(3):656–668. doi: 10.1016/j.molonc.2014.01.008
- Menghi F, Banda K, Kumar P, et al. Genomic and epigenomic. Sci Transl Med. [2022 Jul 6];14(652):eabn1926. doi: 10.1126/scitranslmed.abn1926
- Fernandez SV, Huang Y, Snider KE, et al. Expression and DNA methylation changes in human breast epithelial cells after bisphenol a exposure. Int J Oncol. 2012 Jul;41(1):369–377. doi: 10.3892/ijo.2012.1444
- Al-Yousef N, Shinwari Z, Al-Shahrani B, et al. Curcumin induces re‑expression of BRCA1 and suppression of γ synuclein by modulating DNA promoter methylation in breast cancer cell lines. Oncol Rep. 2020 Mar;43(3):827–838. doi: 10.3892/or.2020.7473