
ABSTRACT
Background: Small monomeric GTPases act as molecular switches in several processes that involve polar cell growth, participating mainly in vesicle trafficking and cytoskeleton rearrangements. This gene superfamily has largely expanded in plants through evolution as compared with other Kingdoms, leading to the suggestion that members of each subfamily might have acquired new functions associated to plant-specific processes. Legume plants engage in a nitrogen-fixing symbiotic interaction with rhizobia in a process that involves polar growth processes associated with the infection throughout the root hair. To get insight into the evolution of small GTPases associated with this process, we use a comparative genomic approach to establish differences in the Ras GTPase superfamily between legume and non-legume plants.
Results: Phylogenetic analyses did not show clear differences in the organization of the different subfamilies of small GTPases between plants that engage or not in nodule symbiosis. Protein alignments revealed a strong conservation at the sequence level of small GTPases previously linked to nodulation by functional genetics. Interestingly, one Rab and three Rop proteins showed conserved amino acid substitutions in legumes, but these changes do not alter the predicted conformational structure of these proteins. Although the steady-state levels of most small GTPases do not change in response to rhizobia, we identified a subset of Rab, Rop and Arf genes whose transcript levels are modulated during the symbiotic interaction, including their spatial distribution along the indeterminate nodule.
Conclusions: This study provides a comprehensive study of the small GTPase superfamily in several plant species. The genetic program associated to root nodule symbiosis includes small GTPases to fulfill specific functions during infection and formation of the symbiosomes. These GTPases seems to have been recruited from members that were already present in common ancestors with plants as distant as monocots since we failed to detect asymmetric evolution in any of the subfamily trees. Expression analyses identified a number of legume members that can have undergone neo- or sub-functionalization associated to the spatio-temporal transcriptional control during the onset of the symbiotic interaction.
Background
Most legume plants have the capacity to establish an intracellular symbiotic interaction with nitrogen-fixing soil bacteria called rhizobia. In this association, bacteria are accommodated inside the cell, surrounded by a host-derived membrane in a subcellular structure specialized in nitrogen fixation. This interaction, known as root nodule symbiosis (RNS), is present only in legumes, with the exception of a small tropical tree of the genus Parasponia.Citation1 Some of the molecular components required to establish this symbiotic interaction apparently evolved by recruitment of pre-existing components from a more ancient interaction between plants and fungi from the phylum Glomeromycota, known as arbuscular mycorrhiza symbiosis (AMS).Citation2,3 In this interaction, fungi are accommodated inside the root cortical cells in structures called arbuscules. The genetic program shared between RNS and AMS is known as the common Sym pathway and its components participate mainly in the molecular changes that occur during infection. On the other hand, RNS involves the formation of a new organ, the nodule, whereas there is not an organogenesis counterpart in AMS. In addition, the recognition of signals derived from the bacteria or the fungi in these two interactions is mediated by different receptors (with the exception of Parasponia), whereas some of the transcription factors required for the establishment of the interaction are also specific for each type of symbiosis. In RNS, the most common infection mechanism begins with the redirectioning of the polar growth of the root hair, which curls to surround the bacterial microcolony, forming an infection focus.Citation4 From this point, a tubular structure, known as the infection thread, grows inward the root hair, guiding the bacteria through the epidermis towards the dividing cortical cells, where bacteria will be released, surrounded by a host-derived membrane and differentiated to bacteroids to form a structure called the symbiosome.Citation5,6 Several molecular components associated with bacterial infection and release have been identified in the last years, including vesicle trafficking components,Citation7-12 cytoskeleton related-proteins.Citation13-16 and cell-wall degrading enzymes.Citation17 Among proteins involved in vesicle trafficking are the small GTPases, some of which play critical roles in early events of the infection process and in the signaling pathway required for nodulation,Citation9-11 but also at later stages during symbiosome maturation.Citation7,8 In a more general context, small GTPases act as molecular binary switches, fluctuating between an activated state when they are bound to GTP and an inactive state bound to GDP.Citation18 They participate in multiple cellular processes across all eukaryotic organisms, being one of the most important groups of regulatory proteins. The superfamily of small GTPases has been divided into five families based on sequence identity.Citation19 Ras GTPases were the first members identified in viruses that can lead to cancer (hence the name Rat sarcoma),Citation20 giving origin to the name that has been adopted for the family and sometimes for the superfamily as well. Ras proteins are part of the signal transduction pathways that regulate cell proliferation in mammals and yeast, but they are absent from plant genomes.Citation21 Proteins of the two other families, Rab (Ras-related in brain) and Arf (ADP ribosylation factor), are involved in the four steps of vesicle trafficking in eukaryotic cells: budding of vesicles, transport, tethering and membrane fusion.Citation22,23 Members of the Rho (Ras homologous) family, known as Rop in plants (Rho of plants), participate in cytoskeleton dynamics and remodeling in response to external stimuli.Citation24 Finally, members of the Ran (Ras-related nuclear protein) family regulate nuclear-cytoplasmic trafficking in both directions across the nuclear pore.Citation25
In the last years, advances in sequencing techniques have facilitated the generation of genomic and transcriptomic datasets, allowing the comparison between species in order to infer the evolutionary history of genomes. The limitation of the RNS to a group of phylogenetically related species, mainly legumes, provides a good opportunity to use comparative genomics and meta-analysis of transcriptomes to understand how different components of the genetic programs have evolved from common ancestors with non-nodulating species, giving origin to new functions in the context of this new biological process. We focused on the small GTPase superfamily, whose members seem to have evolved from functioning in other biological processes to acquire specific functions during the infection process initiated by rhizobia.Citation26 Based on a previous analysis of expressed sequence tags (ESTs), it was suggested that some clades of the Ras superfamily of small GTPases have expanded and undergone neofunctionalization in legumes as a consequence of the new functions required for the establishment of the nitrogen-fixing symbiosis.Citation27,28 In order to re-evaluate this hypothesis, we took advantage of the genomic and transcriptomic datasets recently generated in different species to conduct a comprehensive comparison of the small GTPase gene families and expression patterns of their members in nodulating and non-nodulating species. Our results support the notion that sub- or neo-functionalization are not associated with asymmetric evolution of the gene families or subfamilies, but with the spatio-temporal regulation of gene expression of specific members during the establishment of the symbiotic interaction.
Results and discussion
Phylogenetic analysis of small GTPases in legume and non-legume plants
In order to perform a comprehensive comparison of small GTPases subfamilies between legume and non-legume plants, we selected a group of species whose genomes have been sequenced and transcriptomic datasets are publicly available. Among legumes, the two model plants Lotus japonicus and Medicago truncatula (both associated with forage species) and the two grain legumes common bean (Phaseolus vulgaris) and soybean (Glycine max) were selected. As representatives of non-legumes we included the dicotyledonous Arabidopsis thaliana and tomato (Solanum lycopersicum) and the monocotyledonous rice (Oryza sativa) and maize (Zea mays). Members of the small GTPases superfamily were identified through tBLASTn searches using the amino acid sequence of the monomeric GTPases annotated and classified in Arabidopsis.Citation21-29 as queries and the genome database of each species. Amino acid sequences of these proteins were used to generate phylogenetic trees, allowing their classification in different families and subfamilies ( and Figs. S1-8). As previously described in Arabidopsis, members of Rab, Arf, Rop and Ran families were identified in all species; however, genes encoding proteins of the Ras family were not identified in any of the plant genomes explored in this work (Figs. S1-8).Citation21 No significant differences in the number of members of each family were observed between legume and non-legume species (). The higher number of soybean GTPases compared with other species is most likely related to the partially diploidized tetraploid nature of its genome.Citation30 As each family is subdivided into subfamilies in Arabidopsis, we classified small GTPases from the other selected species using phylogenetic trees that included Arabidopsis members that are representative of each subfamily (Figs. S9-24). Again, we did not observe appreciable differences in the number of genes constituting each subfamily of the monomeric GTPases between the two groups of plants, legume and non-legume species ( & ). Taken together, our phylogenetic analysis in different species did not reveal asymmetric evolution of the small GTPase subfamilies in legume plants associated with the evolution of nitrogen-fixing symbiosis.
Figure 1. Phylogenetic analysis of the small GTPase superfamily in legume and non-legume plants. Amino acid sequences corresponding to small GTPases from A thaliana, O. sativa, S. lycopersicum, Z. mays, L. japonicus, M. truncatula, P. vulgaris, and G. max were retrieved from genomic databases. Unrooted neighbor-joining trees were obtained using the Mega 7 software. Subfamilies were identified for each species: Rab (green), Arf (orange), Rop (blue) and Ran (pink).
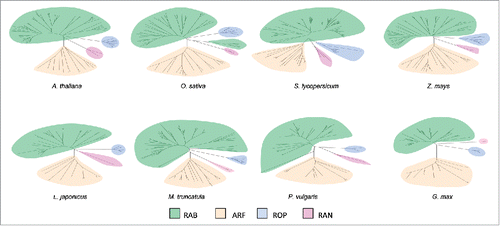
Table 1. Number of members of each small GTPases subfamily.
Table 2. Number of members of the small GTPases Rab subfamily.
Table 3. Number of members of the small GTPases Arf subfamily.
Small GTPases involved in nodulation are structurally conserved among legume and non-legumes
We next wondered whether differences in the amino acid sequence could be linked to specialized functions of small GTPases in the context of physiological changes that occur during rhizobia infection. As described in the introduction, several small GTPases participate in the establishment of symbiosis between legumes and rhizobia. These proteins were used to identify members from other species that were evolutionary related based on phylogenetic analysis (Figs. S25-S29) and sequence identity. We selected PvRabA2 from common bean, MtRab7 (corresponding to a Rab of the G3 group according to the nomenclature suggested by Vernoud et al., 2003,Citation21 MtRop9 and MtRop10 from M. truncatula and LjRop6 from L. japonicus. ArfA1 was also included considering its function in root growthCitation31 and RNS (Dalla Via and Blanco, unpublished results). Multiple sequence alignments were generated for each of these proteins ( and Figs. S30-32). All alignments showed strong amino acid sequence conservation across the different species analyzed here, suggesting that Rab, Arf and Rop proteins were subjected to strong selective pressure. No differences in amino acid sequences were observed in ArfA1 (Fig. S30) and Rab7 (Fig. S31) that could be correlated with legume and non-legume species. PvRabA2 and their putative ortholog genes contain a conserved substitution: whereas a valine residue is in the position 177 in the four legumes, isoleucine is present in that position in the four non-legume species (). Interestingly, three positions show no conservative substitutions in ROP9 proteins (amino acids 53, 129 and 151 are isoleucine, cysteine and asparagine in legumes and threonine, phenylalanine and glycine in non-legumes, respectively). The first substitution is at the end of the effector domain (domain II), whereas the second one affects the RHO insert region (domain V) (). Rop6 is in the same clade that MtRop9 and their amino acid sequences are very similar, but it shows the same single substitution in position 151 that was observed for Rop9 (Fig. S32). Rop10 also contains substitutions in legume species (), but in this case all changes are conservative and only the substitution of lysine in non-legumes by arginine in legumes affects a conserved region.
Figure 2. Multiple sequence alignment of P. vulgaris RABA2 (Phvul.011G061100) and proteins with the highest sequence identity from M. truncatula (Medtr4g064897), G. max (Glyma11g14360), L. japonicus (chr3.CM0792.300.r2.d), A. thaliana (At1g07410), S. lycopersicum (Solyc06g076450), Z. mays (GRMZM2G473906) and O. sativa (LOC_Os03g60870). Black boxes indicate identical residues and gray ones indicate conservative substitutions. Alignments were generated with Clustal Omega in MEGA7 and formatted with Boxshade. The red arrow indicates a conservative amino acid substitution in legume versus non-legume sequences. The conserved domains of Rabs are indicated by blue lines.

Figure 3. Multiple sequence alignment of M. truncatula ROP9 (Medtr5g022600) and proteins with the highest sequence identity from, P. vulgaris (Phvul.002G106600), G. max (Glyma01g36880), L. japonicus ROP6 (chr2.CM0272.860.r2.m), A. thaliana (At2g17800), S. lycopersicum (Solyc02g083580), Z. mays (GRMZM2G375002) and O. sativa (LOC_Os02g58730). Black boxes indicate identical residues and gray ones indicate conservative substitutions. Alignments were generated with Clustal Omega in MEGA7 and formatted with Boxshade. Red arrows indicate amino acid substitutions in legumes versus non-legumes. The conserved domains of ROPs are indicated by blue lines.

Figure 4. Multiple sequence alignment of M. truncatula ROP10 (Medtr3g078260) and proteins with the highest sequence identity from P. vulgaris (Phvul.009G180800), G. max (Glyma04g35110), L. japonicus (chr1.CM0166.830.r2.m), A. thaliana (At3g48040), S. lycopersicum (Solyc03g114070), Z. mays (GRMZM2G415327) and O. sativa (LOC_Os02g50860). Black boxes indicate identical residues and gray ones indicate conservative substitutions. Alignments were generated with Clustal Omega in MEGA7 and formatted with Boxshade. Red arrows indicate amino acid substitutions in legumes versus non-legumes. The conserved domains of ROPs are indicated by blue lines. The sequence from Z. mays is truncated at its C terminus.

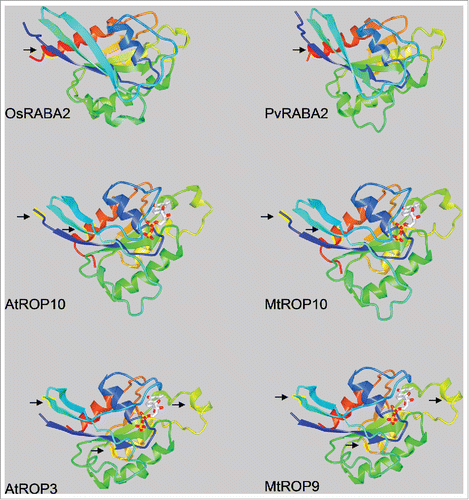
Since the conservation of amino acids across the eight studied species -including monocots and dicots- is an indication of a strong selective pressure, variation of some of these conserved residues in LjRop6, MtRop10, MtRop9 and PvRabA2 in legumes might reflect specialized functions of these proteins in the symbiotic interaction of legumes with rhizobia. However, the comparison of the predicted three dimensional conformations of RabA2, ROP9 and ROP10 suggests that the structure of the proteins is not strongly modified by these substitutions (). Nevertheless, functional analysis would be required to determine if the observed polymorphisms at the sequence level are associated with neo- or sub-functionalization in legume plants.
Figure 5. Three dimensional models of RABA2 from O. sativa and P. vulgaris, ROP10 from A. thaliana and M. truncatula, ROP3 from A. thaliana and ROP9 from M. truncatula. Arrows and yellow boxes indicate the position of the substitutions observed in legume versus non-legume species.
Expression of small GTPases in different tissues
Another possible difference between proteins of the small GTPase families among different species is the spatio-temporal control of gene expression of their members. Using publicly available datasets for each species, we retrieved expression data of each member of small GTPase families (Additional files 2–9; Tables S1-8). Values were normalized using the root sample as a reference and compared to other tissues. Considering the diversity of tissues selected in each study and the biological conditions and/or treatments applied, we selected samples that could be systematically compared among the eight species. In all species, legumes and non-legumes, GTPases of the four families were mainly accumulated at higher levels in roots as compared with aerial tissue (stem and leaf were considered in most cases) or had similar levels in different tissues (Additional files 2–9; Tables S1-8). With the exception of some Rops from common bean and soybean, very few small GTPases showed higher levels of mRNA in aerial tissue as compared with roots (). The distribution of small GTPases with differential expression in root or aerial tissue was not associated with a particular group of Rabs, Arfs or Rops in Arabidopsis (Fig. S1). The spatial expression pattern of small GTPases points toward predominant specific roles in polar and transport processes specific of the root, but the comparison of legume and non-legume plants suggests that these roles are present in all species and probably involve molecular mechanisms that have evolved before the legume family diverged from other groups of plants.
Table 4. Number of each subfamily members that shows no changes (0), reduced (-) or increased (+) levels of mRNA in aerial tissue compared with the root.
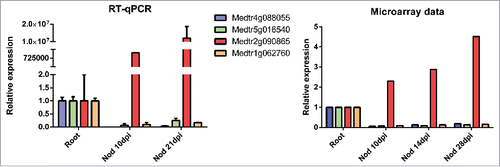
Next, we analyzed the expression of small GTPases in nodules of the four legume species. A small number of Rab and Arf members accumulated at higher levels in nodules as compared to roots. This number was comparable among the four legumes, but lower than the number of small GTPases that exhibited higher transcript levels in roots than in nodules. A similar pattern was observed in Rop mRNA accumulation (). The small GTPases that were up- or down-regulated in roots as compared to nodules do not cluster together in any particular branch of the phylogenetic trees of L. japonicus, M. truncatula or P. vulgaris (Figs. S5-7), suggesting that regulation of their expression in response to rhizobia has evolved independently in different members of each gene family. To validate the expression of a group of selected small GTPases we conducted reverse transcription reactions followed by quantitative PCR (RT-qPCR) in M. truncatula roots and nodules. We selected one ROP (Medtr4g088055), one Arf (Medtr5g016540) and two Rab proteins belonging to the A and C families (Medtr2g090865 and Medtr1g062760, respectively). Consistent with the microarray data, our RT-qPCR data showed that mRNA levels of these genes were higher in roots than in nodules for Medtr4g088055, Medtr5g016540 and Medtr1g062760, whereas an opposite expression pattern was observed for Medtr2g090865 (). These results showed that the expression data obtain by high throughput analysis could be reproduced using a more reliable technique for all the genes analyzed in M. truncatula.
Table 5. Number of each subfamily members that shows no changes (0), reduced (-) or increased (+) levels of mRNA in nodule tissue compared with the root.
Figure 6. Comparison of expression data obtained by RT-qPCR with microarray data of selected M. truncatula genes encoding small GTPases. Expression levels of the four transcripts in roots and nodules (Nod) at 10 or 21 days post inoculation (dpi) with S. meliloti were measured by RT-qPCR or by microarray analysis.Citation48 Bars in qPCR graphs represent media and SE of two biological replicates. Expression levels were normalized with HIS3L and presented relative to the values of root tissue, which was set at 1.
Considering that infections are initiated in the root, before the cortical divisions give origin to the nodule, we compared inoculated with uninoculated roots at very early stages of the interaction (i.e., short time periods after rhizobia inoculation). Surprisingly, only Phvul.004G011900, one member of the Arf family showed a significant difference in the expression level in common bean, with higher transcript levels at 24 hours post-inoculation with rhizobia (Additional file 8; Table S7). Likewise, none of the small GTPases was significantly regulated in roots of L. japonicus at 24 hours after infection with rhizobia (Additional file 6; Table S5) or in M. truncatula root hairs at early stages of the interaction (1, 3 and 5 dpi) when roots inoculated with a wild type strain were compared with roots inoculated with a non-fixing strain of Sinorrhizobium melilotiCitation32 (Additional file 7; Table S6). These results suggest that, besides the biological important functions played by the small GTPases during early stages of legume-rhizobia symbiosis, these molecular switches are not strongly regulated at transcriptional level and/or at the level of their mRNA stability.
Spatial distribution of small GTPases transcripts in indeterminate nodules
Indeterminate nodules are divided into developmental zones according to the specific functions played by the different cell types found within the organ.Citation6 We took advantage of the laser-capture microdissection strategy applied to dissect the transcriptome of different zones of M. truncatula indeterminate nodules to characterize the spatial distribution of transcripts encoding each member of the small GTPase family.Citation33 In that study, different zones from the mature nodule were excised using a laser microdissection microscope and RNA from each nodule zone was subjected to transcriptome analysis. Based on clustering analysis, 13 expression patterns were defined according to mRNA abundance in the meristematic (FI), the infection zone (FII), the interzone (IZ) or the fixation zone (FIII). These data allow us to study the transcript levels of individual members of the small GTPase family in the context of the nodule organogenesis (Additional file 10; Table S9). Several members of Rab, Arf, Rop and Ran subfamilies showed differential accumulation of mRNA across the different nodule zones (). Clusters 1 to 5 showed a high relative transcript abundance in the meristematic and infection zones, whereas clusters 6, 11, 12 exhibited higher expression around the fixation zone. Interestingly, four Rab genes were grouped in cluster 1 and 2, showing a strong expression in the meristem (Medtr8g027220) or the meristem and the pre-infection zone (FIId; Medtr2g075950, Medtr3g086980 and Medtr7g051940). Only one Rop showed enhanced expression in the meristem (Medtr6g087980), whereas no Arf proteins were grouped in these clusters. Expression patterns of these genes suggest that the encoded small GTPase can play roles in nodule meristem activity/maintenance or at very early events of the infection process. On the other hand, several Rabs, Rops and Arf were grouped in the clusters 11 and 12, showing higher accumulation of their mRNAs in the fixation zone, where bacteria are already differentiated inside the symbiosomes (). Five Rabs and one Arf were included in cluster 13, which includes genes that are expressed in all nodule zones, but preferentially in the fixation zone. GTPases encoded by genes from cluster 11, 12 and 13 could be involved in the maintenance of symbiosome membrane identity or the exchange of metabolites across the symbiosome membrane between the macro- and the micro-symbionts. Taken together, these results revealed that the expression of distinct small GTPases is differentially regulated at the spatial level in nodule zones that exert highly specialized functions during root nodule symbiosis and suggest that symbiotic functions of these proteins can be associated with their expression patterns.
Table 6. Expression of M. truncatula GTPases in different regions of the mature noduleFootnotea.
Concluding remarks
The development of comparative genomics over the past years has contributed to understand functionalization of genes in an evolutionary context. In this work, we aimed to unveil how members of the small GTPase superfamily have acquired specific functions in the legume family associated with the unique capacity to establish RNS. Our analyses suggest that the number of family members and the primary sequence of small GTPases are well conserved between legume and non-legume plants. More importantly, small GTPases that have been linked to the symbiotic association by genetic approaches have highly conserved homolog genes in non-legume plants. However, we were able to identify specific amino acid residues in some of these GTPases that have changed during the course of evolution, beside the extreme amino acid conservation among genetically distant species. It will be of great interest to explore the functional relevance of these substitutions in the context of the symbiotic interaction established between legumes and rhizobia. Another interesting aspect of our analysis is the identification of specific members of each of the Rab, Arf and Rop families that show changes in the steady-state level of their mRNAs during the onset of the symbiotic interaction. Spatial information provided by laser-capture microdissection allowed us to identify several members that are also regulated along the different zones of the indeterminate nodule, suggesting that these small GTPases could have acquired specific functions that operate during infection, progression of the IT, symbiosome release or allocation and maintenance of nitrogen-fixing symbiosomes. In addition to provide insight into these interesting aspects of the symbiotic interaction, we believe this study presents a comprehensive compendium of a relevant and complex gene family in eight different species, establishing the basis for the study of the diverse biological processes in which small GTPases could be involved.
Methods
Identification of small GTPases from different species
Members of the small GTPases superfamily were identified through tBLASTn searchesCitation34 using the amino acid sequence of all members of the small GTPase family previously described and classified in ArabidopsisCitation21,29 as queries and the genomic dataset of each species (see Availability of data and materials). The list of identified genes were manually curated and classified according to our phylogenetic analyses (see below).
Phylogenetic analysis
Amino acid sequences were aligned using Clustal Omega http://www.ebi.ac.uk/Tools/msa/clustaloCitation35 and the phylogenetic analysis was performed with the MEGA7 packageCitation36 http://www.megasoftware.net using the neighbor-joining methodCitation37 with 1000 trials to obtain bootstrap values. The evolutionary distances were computed using the number of differences method.Citation38 All positions containing gaps and missing data were eliminated from the dataset. The phylogenetic analysis is available in Figs. S1-29. For Figs. S25-29, the branches with bootstrap values lower than 29% were collapsed to simplify the trees. Alignment files are provided in Additional file 11.
Protein alignments
The sequences of small GTPases involved in the establishment of symbiosis between legumes and rhizobia were used to identify members from other species using BLASTP. We selected PvRabA2 from common bean, MtRab7, MtRop9 and MtRop10 from M. truncatula and LjRop6 from L. japonicus and PvArfA1 as queries and the genome database of each species. The amino acid sequences with the lowest E value were used to generate a multiple sequence alignment.
Alignments were generated with Clustal Omega (http://www.ebi.ac.uk/Tools/msa/clustalo) in MEGA7 (http://www.megasoftware.net) and formatted with Boxshade (http://embnet.vital-it.ch/software/BOX_form.html).
3D modeling
Proteins were modeled using Swiss Model.Citation39 (https://swissmodel.expasy.org/) and analyzed using the 3D structure viewer iCn3D (https://www.ncbi.nlm.nih.gov/Structure/icn3d/full.html).
Genomic datasets
Sequences were obtained from public datasets: Arabidopsis thaliana TAIR10, Oryza sativa v7_JGI, Zea mays Ensembl-18, Solanum lycopersicum iTAG2.3, Medicago truncatula Mt4.0v1. Glycine max Wm82.a2.v1 and Phaseolus vulgaris v1.0 are available at Phytozome v11.0 (https://phytozome.jgi.doe.gov/pz/portal.html) and Lotus japonicus v2.5, is available in Miyakogusa v2.5 (http://www.kazusa.or.jp/lotus).
Transcriptomic datasets
Gene expression of small GTPases was explored using publicly available databases.
- A. thaliana: expression data were obtained from the Transcriptome Variation Analysis database (TraVA, http://travadb.org.Citation40 That study analyzed RNA-seq data from 79 samples, each with two biological replicates, corresponding to different developmental stages and parts of roots, leaves, flowers, seeds, siliques and stems from A. thaliana ecotype Col-0. Differential gene expression analysis between all possible pairs of samples were performed using DESeq.Citation41 A false discovery rate (FDR) of 0.05 and a fold change of 2.0 were chosen as the threshold for significantly differential expression.
- S. lycopersicum expression data were obtained from the Tomato Functional Genomics Database (http://ted.bti.cornell.edu).Citation42 That study contains Illumina RNA-seq data from root, stem, leaf, flower and fruit at 3 different maturation stages: mature green, breaker and ripe of tomato cultivar Heinz. Two biological replicates were analyzed. Differentially expressed genes were identified using DESeq and raw p values of multiple tests were corrected using FDR.
- O. sativa: expression data were retrieved from.Citation43 That study includes gene expression data obtained using Illumina RNAseq technology from rice roots and shoots in different stages of Pi deprivation. Three biological replicates of Oriza sativa cv Nipponbare were used. Identification of differentially expressed genes was performed using CuffdiffCitation44 to analyze the effects of Pi status on the transcriptome of roots and shoots. Additional expression data were obtained from,Citation45 which contains RNAseq data from nine distinct tissues including immature leaves, pre and post-emergence flowers, anther, pistil, whole seed at two different maturation stages, embryo, endosperm and biological replicates of rice cv. Nipponbare.
- Z. mays: expression data were obtained from RNAseq transcriptomic analysesCitation46 that include expression data from 18 tissues representing five organs. Samples were obtained from the reference inbred line B73. Three replicates were used for all the analyses. RNA-Seq expression data were compared with the previously published microarray based gene atlas.Citation47
- M. truncatula: data from The Medicago truncatula Gene Expression Atlas MtGEA (http://mtgea.noble.org/v3,Citation48) was used. The study was performed using the Affymetrix GeneChip Medicago Genome Array. Gene expression values were obtained from three independent biological replicates of each organ: roots, nodules, stems, petioles, leaf blades, vegetative buds, flowers, and seed pods of M. truncatula cv Jemalong A17. In addition, three stages of nodule and six stages of seed development were profiled. Hierarchical clustering analysis was conducted with Spotfire DecisionSite 8.1 (Spotfire Inc., http://spotfire.tibco.com/). For the nodule developmental series, transcript levels were expressed relative to the level in roots just prior to inoculation and clustered using the Pearson correlation coefficient. Additionally, we explored the transcriptome data of the laser-capture microdissection strategy applied to M. truncatula indeterminate nodulesCitation33 to characterize of gene expression of the small GTPase family in different nodule zones. In addition, whole roots, whole nodules and aerial organs were profiled. Three biological repetitions were analyzed in all cases except for pooled aerial organs (one experiment). Based on clustering analysis, 13 expression patterns were defined according to mRNA abundance in the meristematic (FI), the infection zone (FII), the interzone (IZ) or the fixation zone (FIII). LCM methodology was validated by Roux et al with a set of 37 marker genes previously characterized by other methods.Citation33 Additional expression data were obtained from root hairs prior to and during the initial stages of infection with S. meliloti.Citation32 Three biological replicates were performed for each treatment. Wild-type (Jemalong A17) or skl-1 mutant plants were used at different days post inoculation with Sinorhizobium meliloti or a S. meliloti non-fixing strain. A17 was also profiled at 1 d post-treatment following addition of Nod factors.
- L. japonicus: data were obtained from The Lotus japonicus Gene Expression Atlas LjGEA (http://ljgea.noble.org/v2,Citation49). This work presents an integrated genome-wide analysis of transcriptome landscapes of wild-type (ecotype Gifu) and symbiotic mutant plants using the Affymetrix GeneChip Lotus Array. Five different organs, five stages of the sequentially developed determinate root nodules, and eight mutants impaired at different stages of the symbiotic interaction were analyzed in triplicates from three spatially and temporally separated batches of plants. Significant genes were identified using the Limma package.Citation50 A FDR (corrected p-value < 0.05) was used as the criterion for significance, in combination with a |M|≥1 filter, where M is the log2 ratio of average expression values from any two conditions.
- P. vulgaris: expression data were obtained from the Common Bean Gene Expression Atlas (http://plantgrn.noble.org/PvGEA,Citation51), which presents gene expression values obtained by RNAseq from 24 unique samples collected from seven distinct tissues of P. vulgaris cv. Negro Jamapa; roots, nodules, leaves, stems, flowers, seeds, and pods. Plants were inoculated with either effective or ineffective strains of rhizobia. Transcripts differentially expressed between libraries were identified using NOIseq.Citation52 In that study, differential expression was confirmed by reverse transcription followed by quantitative PCR (RT-qPCR) analyses for 92% of the 85 genes selected. Additional expression data were retrieved from our transcriptomic analysis of P. vulgaris cv. NAG12 plants inoculated with different Rhizobium etli strains at 24 hours.Citation53 Two biological replicates were used for RNAseq and differentially expressed genes were identified using Cuffdiff. RT-qPCR analysis of differentially expressed genes confirmed expression data obtained by RNAseq for nine out of 11 genes.Citation53
- Glycine max: data were obtained from the SoyBase Database (https://www.soybase.org/soyseq,Citation54) which includes RNA-Seq from seven tissues (leaf, flower, pod, two stages of pod-shell, root, nodule) and seven stages in seed development. General trends in expression profiles for all genes were examined by a comparison of the transcription count for every tissue to every other tissue using a Fisher 's exact test with a FDR correction of 0.05.
Expression data for all small GTPase members from each species are available in Additional files 2–10, Tables S1-9. The Join two Datasets tool available at Galaxy platformCitation55 was used to merge expression data from different datasets.
Validation of Expression Data by qRT-PCR
We selected a set of four differentially expressed genes corresponding to small GTPases (Medtr2g090865, Medtr5g016540, Medtr4g088055 and Medtr1g062760) to validate their expression using RT-qPCR and to compare with that previously obtained by microarray by Benedito et al.Citation48 RT-qPCR was performed essentially as previously described.Citation56 For each pair of primers (Additional file 12, Table S10), the presence of a unique PCR product of the expected size was verified in ethidium bromide-stained agarose gels. Absence of contaminant genomic DNA was confirmed in reactions with DNAse-treated RNA as template. Expression values were normalized to HIS3L, which has been validated by GNORM.Citation57 Two biological replicates were performed.
Statistical analysis of expression data
Using publicly available datasets for each species, we retrieved expression data of each member of small GTPases families. We selected roots as the reference organ and tested their expression in other organs for the eight species. The expression values were normalized using the root sample and the log2 (fold change) was calculated for each tissue. For A. thaliana and G. max, genes were considered as differentially expressed according to the statistical analyses performed by the authors.Citation40,Citation54 We analyzed the data retrieved fromCitation43 for O. sativa and data from common bean using CuffDiffCitation44 to identify genes differentially expressed between root and shoot. Genes whose transcript levels showed at least a 2-fold change between samples (log2 fold change <-1 or >1), a p value < 0.05 and an expression > 1 fragment per kilobase per million (FPKM) in at least one sample were considered as differentials.
For the others species, genes assigned with reduced (-) or increased (+) transcript level were those that exhibited at least a log2 fold change <-1 or >1, respectively. Transcripts with -1<log2 fold change<1 were considered as genes with no changes in gene expression (0). Only values with FPKM >1 or reads per kilobase per million reads (RPKM) >1 in at least one sample were considered.
In order to analyze the expression of small GTPases in the nodules of the four legume species, we compared their expression to root samples. For M. truncatula and G. max, the genes considered as differentially expressed were assigned according to the statistical analyses from the authors.Citation48,Citation54 For L. japonicus and P. vulgaris, differentially expressed transcripts were those with at least a 2-fold change between root tissue and at least one sample of nodule tissue (log2 fold change >1 or <-1).
Abbreviations
| 3D | = | Three dimensional |
| AMS | = | arbuscular mycorrhiza symbiosis |
| ARF | = | ADP ribosylation factor |
| EST | = | expressed sequence tag |
| GDP | = | Guanosine-5'-diphosphate |
| GTP | = | Guanosine-5'-triphosphate |
| NFR5 | = | Nod Factor Receptor 5 |
| RAB | = | Ras-related in brain |
| RAN | = | Ras-related nuclear protein |
| RAS | = | Rat sarcoma |
| RHO | = | Ras homologous |
| ROP | = | Rho of plants |
| RNS | = | root nodule symbiosis |
| vs | = | versus |
Declarations
Ethics approval and consent to participate: Not applicable
Consent for publication: Not applicable
Availability of data and materials:
Genomic datasets
Sequences were obtained from public datasets: Arabidopsis thaliana TAIR10, Oryza sativa v7_JGI, Zea mays Ensembl-18, Solanum lycopersicum iTAG2.3, Medicago truncatula Mt4.0v1, Glycine max Wm82.a2.v1 and Phaseolus vulgaris v1.0 are available on the Phytozome v11.0 (https://phytozome.jgi.doe.gov/pz/portal.html) and Lotus japonicus v2.5, is available on Miyakogusa v2.5 (http://www.kazusa.or.jp/lotus).
Transcriptomic datasets
Gene expression atlases were explored in order to characterize the transcriptional patterns of the A. thaliana (TraVA, Transcriptome Variation Analysis, http://travadb.org,Citation40), S. lycopersicum (Tomato Functional Genomics Database http://ted.bti.cornell.edu,Citation42), M. truncatula (The Medicago truncatula Gene Expression Atlas MtGEA http://mtgea.noble.org/v3,Citation48), L. japonicus (The Lotus japonicus Gene Expression Atlas LjGEA http://ljgea.noble.org/v2,Citation49), P. vulgaris (A Common Bean Gene Expression Atlas http://plantgrn.noble.org/PvGEA,Citation51) and Glicine max (SoyBase Database https://www.soybase.org/soyseq.Citation54) Additional expression data were retrieved for O. sativa.Citation43 Z. mays,,Citation46 P. vulgaris.Citation53 and M. truncatula.Citation32,33 The expression data for each species is available in Additional files 2–10; Tables S1-9.
Competing interest
The authors declare that they have no competing interests.
Authors' contributions
FAB design research; AF, VDV, VS and UM collected data and performed the analysis. AF, MEZ and FAB wrote the article.
Suppl_mat_Comparative_phylogenetic_and_expression_analysis_of_small_Gtpases_families.zip
Download Zip (16.1 MB)Acknowledgments
We thank Claudio Rivero and Soledad Traubenik for assistance with Tables and Carla Roda and Soledad Traubenik for reading the manuscript.
Funding
This work was financially supported by grants from ANPCyT, Argentina (PICT 2013/0384 and 2014/0321). VS, UMV, MEZ and FAB are funded by CONICET and ACF is funded by UNLP.
Related Research Data
References
- Op den Camp R, Streng A, De Mita S, Cao Q, Polone E, Liu W, Ammiraju JS, Kudrna D, Wing R, Untergasser A, et al. LysM-Type Mycorrhizal Receptor Recruited for Rhizobium Symbiosis in Nonlegume Parasponia. Science. 2011;331:909–912. doi:10.1126/science.1198181.
- Kistner C, Parniske M. Evolution of signal transduction in intracellular symbiosis. Trends Plant Sci. 2002;7(11):511–518. doi:10.1016/S1360-1385(02)02356-7.
- Markmann K, Parniske M. Evolution of root endosymbiosis with bacteria: How novel are nodules? Trends Plant Sci. 2009;14(2):77–86. doi:10.1016/j.tplants.2008.11.009.
- Oldroyd GE, Murray JD, Poole PS, Downie JA. The rules of engagement in the legume-rhizobial symbiosis. Annu Rev Genet. 2011;45:119–44. doi:10.1146/annurev-genet-110410-132549.
- van Brussel AA, Bakhuizen R, van Spronsen PC, Spaink HP, Tak T, Lugtenberg BJ, Kijne JW. Induction of Pre-Infection Thread Structures in the Leguminous Host Plant by Mitogenic Lipo-Oligosaccharides of Rhizobium. Science. 1992;257(5066):70–72. doi:10.1126/science.257.5066.70.
- Timmers AC, Auriac MC, Truchet G. Refined analysis of early symbiotic steps of the Rhizobium-Medicago interaction in relationship with microtubular cytoskeleton rearrangements. Development. 1999;126(16):3617–28.
- Cheon CI, Lee NG, Siddique AB, Bal AK, Verma DP. Roles of plant homologs of Rab1p and Rab7p in the biogenesis of the peribacteroid membrane, a subcellular compartment formed de novo during root nodule symbiosis. EMBO J. 1993;12(11):4125–35.
- Limpens E, Ivanov S, van Esse W, Voets G, Fedorova E, Bisseling T. Medicago N2-Fixing Symbiosomes Acquire the Endocytic Identity Marker Rab7 but Delay the Acquisition of Vacuolar Identity. Plant Cell. 2009;21(9):2811–28. doi:10.1105/tpc.108.064410.
- Blanco FA, Meschini EP, Zanetti ME, Aguilar OM. A small GTPase of the Rab family is required for root hair formation and preinfection stages of the common bean-Rhizobium symbiotic association. Plant Cell. 2009;21(9):2797–2810. doi:10.1105/tpc.108.063420.
- Ke D, Fang Q, Chen C, Zhu H, Chen T, Chang X, Yuan S, Kang H, Ma L, Hong Z, et al. The Small GTPase ROP6 Interacts with NFR5 and Is Involved in Nodule Formation in Lotus japonicus. Plant Physiol. 2012;159(1):131–43. doi:10.1104/pp.112.197269.
- Kiirika LM, Bergmann HF, Schikowsky C, Wimmer D, Korte J, Schmitz U, Niehaus K, Colditz F. Silencing of the Rac1 GTPase MtROP9 in Medicago truncatula Stimulates Early Mycorrhizal and Oomycete Root Colonizations But Negatively Affects Rhizobial Infection. Plant Physiol. 2012;159(1):501–16. doi:10.1104/pp.112.193706.
- Dalla Via V, Traubenik S, Rivero C, Aguilar OM, Zanetti ME, Blanco FA. The monomeric GTPase RabA2 is required for progression and maintenance of membrane integrity of infection threads during root nodule symbiosis. Plant Mol Biol. 2017;93(6):549–62. doi:10.1007/s11103-016-0581-5.
- Yokota K, Fukai E, Madsen LH, Jurkiewicz A, Rueda P, Radutoiu S, Held M, Hossain MS, Szczyglowski K, Morieri G, et al. Rearrangement of Actin Cytoskeleton Mediates Invasion of Lotus japonicus Roots by Mesorhizobium loti. Plant Cell. 2009;21(1):267–84. doi:10.1105/tpc.108.063693.
- Miyahara A, Richens J, Starker C, Morieri G, Smith L, Long S, Downie JA, Oldroyd GED. Conservation in Function of a SCAR/WAVE Component During Infection Thread and Root Hair Growth in Medicago truncatula. Mol Plant-Microbe Interact. 2010;23(12):1553–62. doi:10.1094/MPMI-06-10-0144.
- Fournier J, Teillet A, Chabaud M, Ivanov S, Genre A, Limpens E, de Carvalho-Niebel F, Barker DG. Remodeling of the Infection Chamber before Infection Thread Formation Reveals a Two-Step Mechanism for Rhizobial Entry into the Host Legume Root Hair. Plant Physiol. 2015;167(4):1233–42. doi:10.1104/pp.114.253302.
- Hossain MS, Liao J, James EK, Sato S, Tabata S, Jurkiewicz A, Madsen LH, Stougaard J, Ross L, Szczyglowski K. Lotus japonicus ARPC1 Is Required for Rhizobial Infection. Plant Physiol. 2012;160(2):917–28. doi:10.1104/pp.112.202572.
- Xie F, Murray JD, Kim J, Heckmann AB, Edwards A, Oldroyd GE, Downie JA. Legume pectate lyase required for root infection by rhizobia. Proc Natl Acad Sci U S A. 2012;109(2):633–38. doi:10.1073/pnas.1113992109.
- Takai Y, Sasaki T, Matozaki T. Small GTP-Binding Proteins. Physiological Reviews. 2001;81(1):153–208. doi:10.1152/physrev.2001.81.1.153.
- Kahn RA, Der CJ, Bokoch GM. The ras superfamily of GTP-binding proteins: guidelines on nomenclature. FASEB J. 1992;6(8):2512–13. doi:10.1096/fasebj.6.8.1592203.
- Cooper G. Cellular transforming genes. Science. 1982;217(4562):801–806. doi:10.1126/science.6285471.
- Vernoud V, Horton AC, Yang Z, Nielsen E. Analysis of the small GTPase gene superfamily of Arabidopsis. Plant Physiol. 2003;131(3):1191–208. doi:10.1104/pp.013052.
- Stenmark H, Olkkonen V. The Rab GTPase family. Genome Biology. 2001;2(5):reviews3007.3001. doi:10.1186/gb-2001-2-5-reviews3007.
- Pasqualato S, Renault L, Cherfils J. Arf, Arl, Arp and Sar proteins: A family of GTP-binding proteins with a structural device for ‘front–back’ communication. EMBO Reports. 2002;3(11):1035–41. doi:10.1093/embo-reports/kvf221.
- Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420(6916):629–35. doi:10.1038/nature01148.
- Cavazza T, Vernos I. The RanGTP Pathway: From Nucleo-Cytoplasmic Transport to Spindle Assembly and Beyond. Front Cell Dev Biol. 2016;3(82).
- Rivero C, Traubenik S, Zanetti ME, Blanco FA. Small GTPases in plant biotic interactions. Small GTPases. 2017. doi:10.1080/21541248.2017.1333557.
- Yuksel B, Memon AR. Comparative phylogenetic analysis of small GTP-binding genes of model legume plants and assessment of their roles in root nodules. J Exp Bot. 2008;59(14):3831–3844. doi:10.1093/jxb/ern223.
- Yuksel B, Memon AR. Legume small GTPases and their role in the establishment of symbiotic associations with Rhizobium spp. Plant Signal Behav. 2009;4(4):257–60. doi:10.4161/psb.4.4.7868.
- Rutherford S, Moore I. The Arabidopsis Rab GTPase family: another enigma variation. Curr Opin Plant Biol. 2002;5(6):518–28. doi:10.1016/S1369-5266(02)00307-2.
- Singh RJ, Hymowitz T. The genomic relationship between Glycine max (L.) Merr. and G. soja Sieb. and Zucc. as revealed by pachytene chromosome analysis. Theor App Genet. 1988;76(5):705–11. doi:10.1007/BF00303516.
- Xu J, Scheres B. Dissection of Arabidopsis ADP-RIBOSYLATION FACTOR 1 Function in Epidermal Cell Polarity. Plant Cell. 2005;17(2):525–36. doi:10.1105/tpc.104.028449.
- Breakspear A, Liu C, Roy S, Stacey N, Rogers C, Trick M, Morieri G, Mysore KS, Wen J, Oldroyd GED, et al. The Root Hair “Infectome” of Medicago truncatula Uncovers Changes in Cell Cycle Genes and Reveals a Requirement for Auxin Signaling in Rhizobial Infection. Plant Cell. 2014;26(12):4680–4701. doi:10.1105/tpc.114.133496.
- Roux B, Rodde N, Jardinaud M-F, Timmers T, Sauviac L, Cottret L, Carrère S, Sallet E, Courcelle E, Moreau S, et al. An integrated analysis of plant and bacterial gene expression in symbiotic root nodules using laser-capture microdissection coupled to RNA sequencing. Plant J. 2014;77(6):817–837. doi:10.1111/tpj.12442.
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–402. doi:10.1093/nar/25.17.3389.
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Sys Biol. 2011;7:539–39.
- Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–74. doi:10.1093/molbev/msw054.
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4(4):406–25.
- Nei M, Kumar S. Molecular Evolution and Phylogenetics. Genetical Research. 2001;77(1):117–20.
- Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Cassarino TG, Bertoni M, Bordoli L, et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42(W1):W252–58. doi:10.1093/nar/gku340.
- Klepikova AV, Kasianov AS, Gerasimov ES, Logacheva MD, Penin AA. A high resolution map of the Arabidopsis thaliana developmental transcriptome based on RNA-seq profiling. Plant J. 2016;88(6):1058–70. doi:10.1111/tpj.13312.
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106. doi:10.1186/gb-2010-11-10-r106.
- The Tomato Genome Consortium. The tomato genome sequence provides insights into fleshy fruit evolution. Nature. 2012;485(7400):635–41. doi:10.1038/nature11119.
- Secco D, Jabnoune M, Walker H, Shou H, Wu P, Poirier Y, Whelan J. Spatio-Temporal Transcript Profiling of Rice Roots and Shoots in Response to Phosphate Starvation and Recovery. Plant Cell. 2013;25:4285–304. doi:10.1105/tpc.113.117325.
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protocols. 2012;7(3):562–78. doi:10.1038/nprot.2012.016.
- Davidson RM, Gowda M, Moghe G, Lin H, Vaillancourt B, Shiu S-H, Jiang N, Robin Buell C. Comparative transcriptomics of three Poaceae species reveals patterns of gene expression evolution. Plant J. 2012;71(3):492–502.
- Sekhon RS, Briskine R, Hirsch CN, Myers CL, Springer NM, Buell CR, de Leon N, Kaeppler SM. Maize gene atlas developed by RNA sequencing and comparative evaluation of transcriptomes based on RNA sequencing and microarrays. PLoS ONE. 2013;8(4):e61005. doi:10.1371/journal.pone.0061005.
- Sekhon RS, Lin H, Childs KL, Hansey CN, Buell CR, de Leon N, Kaeppler SM. Genome-wide atlas of transcription during maize development. Plant J. 2011;66(4):553–63. doi:10.1111/j.1365-313X.2011.04527.x.
- Benedito VA, Torres-Jerez I, Murray JD, Andriankaja A, Allen S, Kakar K, Wandrey M, Verdier J, Zuber H, Ott T, et al. A gene expression atlas of the model legume Medicago truncatula. Plant J. 2008;55(3):504–13. doi:10.1111/j.1365-313X.2008.03519.x.
- Høgslund N, Radutoiu S, Krusell L, Voroshilova V, Hannah MA, Goffard N, Sanchez DH, Lippold F, Ott T, Sato S, et al. Dissection of symbiosis and organ development by integrated transcriptome analysis of Lotus japonicus mutant and wild-type plants. PLoS ONE. 2009;4(8):e6556. doi:10.1371/journal.pone.0006556.
- Smyth GK. limma: Linear Models for Microarray Data. In: Bioinformatics and Computational Biology Solutions Using R and Bioconductor. Edited by Gentleman R, Carey VJ, Huber W, Irizarry RA, Dudoit S. New York, NY: Springer New York; 2005:397–420.
- O'Rourke JA, Iniguez LP, Fu F, Bucciarelli B, Miller SS, Jackson SA, McClean PE, Li J, Dai X, Zhao PX, et al. An RNA-Seq based gene expression atlas of the common bean. BMC Genomics. 2014;15(1):866. doi:10.1186/1471-2164-15-866.
- Tarazona S, García-Alcalde F, Dopazo J, Ferrer A, Conesa A. Differential expression in RNA-seq: A matter of depth. Genome Res. 2011;21(12):2213–23. doi:10.1101/gr.124321.111.
- Dalla Via V, Narduzzi C, Aguilar OM, Zanetti ME, Blanco FA. Changes in the common bean (Phaseolus vulgaris) transcriptome in response to secreted and surface signal molecules of Rhizobium etli. Plant Physiol. 2015;169(2):1356–70. doi:10.1104/pp.15.00508.
- Severin AJ, Woody JL, Bolon Y-T, Joseph B, Diers BW, Farmer AD, Muehlbauer GJ, Nelson RT, Grant D, Specht JE, et al. RNA-Seq Atlas of Glycine max: A guide to the soybean transcriptome. BMC Plant Biol. 2010;10(1):160. doi:10.1186/1471-2229-10-160.
- Afgan E, Baker D, van den Beek M, Blankenberg D, Bouvier D, Čech M, Chilton J, Clements D, Coraor N, Eberhard C, et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016;44(Web Server issue):W3–W10. doi:10.1093/nar/gkw343.
- Reynoso MA, Blanco FA, Zanetti ME. Insights into post-transcriptional regulation during legume-rhizobia symbiosis. Plant Signal Behav. 2013;8(2):e23102. doi:10.4161/psb.23102.
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3(7): research0034.0031-research0034.0011. doi:10.1186/gb-2002-3-7-research0034.