
ABSTRACT
Long non-coding RNAs (lncRNAs) are important regulatory factors for plant growth and development. Despite this, little is known about the regulatory interactions of lncRNAs with mRNA during embryo development. Here, we used a bioinformatics genome-wide approach to identify lncRNAs involved in embryo development of Ginkgo biloba, based on RNA sequencing datasets from G. biloba embryos during early, middle, late developmental stages. In total, 2326 lncRNAs were identified in the G. biloba embryos, of which 1307 and 1019 could be classified as long intergenic non-coding RNAs and antisense lncRNAs, respectively. Among them, a total of 657 differentially expressed lncRNAs were identified in the different developmental stages of the G. biloba embryos. Based on the functional annotation of potential target genes of lncRNAs, 50, 33, and 76 lncRNAs were predicted to target genes involved in plant hormone signal transduction, plant hormone biosynthesis, and circadian rhythm regulation, respectively. A lncRNA (17)-miRNA (25)-PCgene (52) network was constructed for the G. biloba embryo. Three lncRNAs (lnc000823, lnc002072, lnc000866) were predicted as target mimics of miR159, which targeted two transcription factors with variety of functions, Gb_11536 (MYB33) and Gb_23921 (MYB101). The data generated in this study provide a better understanding of the roles of lncRNAs in embryo development of G. biloba and plants in general.
Introduction
Long non-coding RNAs (lncRNAs) are transcripts that do not encode proteins and are longer than 200 nucleotides. In recent years, emerging studies have demonstrated that lncRNAs are involved in diverse biological processes, such as DNA methylationCitation1 and chromatin remodeling.Citation2 They can also act as microRNA (miRNA) sponges, increasing the expression of mRNAs targeted by the sequestered miRNA.Citation3 lncRNAs from seeds have been identified in many plants, such as castor bean,Citation1 maize,Citation4 rice,Citation5,Citation6 Brassica napus,Citation7 and tree peony,Citation8 and play crucial roles in seed development, reproductive processes, and lipid biosynthesis. However, little is known about how lncRNAs contribute to embryo development in plants.
Ginkgo biloba seeds have a relatively large embryo compared with other gymnosperms, such as Cunninghamia lanceolata, which have extremely small embryos. Thus, G. biloba embryos are good specimens for studying the development of gymnosperm embryos. Ginkgo seeds mature and fall naturally, prior to complete development of the embryo. The developing embryo then requires a period of post-ripening about 150 days before the seed is able to germinate. Therefore, the G. biloba seed can be described as exhibiting morphophysiological dormancy, which is defined as a dormancy in which embryos need more than 30 days to grow and germinate, and requires a dormancy-breaking treatment.Citation9 During the period of post-ripening, G. biloba embryonic development go through not only morphological changes, such as tissue differentiation and organ formation, the accumulation and metabolism of nutrients and hormone levels will also change. Therefore, G. biloba embryo development plays a key role in seed dormancy and germination. Although much effort has been put to better understand how G. biloba seed germination is controlled by various environmental factors and applied chemicals, the present knowledge based on endogenous signals is still insufficient. It seemed necessary, therefore, to explore when and how the complex regulatory mechanism of morphophysiological dormancy works at the genomic level during the long period of Ginkgo embryo after-ripening till seed germination.
The aim of this study is to identify and characterize lncRNAs from G. biloba embryos to explore genetic differences during Ginkgo embryo development. RNA sequencing (RNA-seq) datasets from early [0–0.3 cm, cotyledons began to swell], middle [0.3–0.7 cm, hypocotyl and cotyledon elongated], and late [> 0.7 cm, radicle elongated but not broke through the testa] developmental stages of G. biloba embryos were used to analyze. lncRNAs that are associated with seed dormancy and lncRNA-miRNA-mRNA networks in Ginkgo embryo development were analyzed. Our findings will help elucidate the potential functions of lncRNAs in Ginkgo embryo development.
Results
Identification and characterization of lncRNAs in Ginkgo embryos
The transcript data underwent stringent filtration to identify lncRNAs from the G. biloba embryos. First, transcripts with a length of less than 200 nucleotides were filtered out, leaving 95,223 transcripts. Then, known protein coding genes (PCgenes) were removed, bringing the dataset down to 3916 transcripts with no overlap to known PCgenes in the sense strand. Next, transcripts with expression levels less than 0.5 were removed, decreasing the dataset size to 3712 transcripts. Finally, the transcripts were subjected to the Coding Potential Calculator (CPC) and Coding-Non-Coding index (CNCI). The final curated dataset included 2326 lncRNA transcripts, with 1307 (56.2%) lincRNAs and 1019 (43.8%) antisense lncRNAs (, Supplementary Table S1).
Figure 1. Characterization of long non-coding RNAs (lncRNAs) in Gingko embryos. (a) Embryo of a Gingko seed. Bar = 0.3 cm; arrow = embryo. (b) Open reading frame length density distribution of lncRNAs and mRNAs. (c) Distribution of exon numbers in lncRNAs and mRNAs. (d) Length density distributions of long intergenic noncoding RNAs (lincRNAs), antisense lncRNAs, and mRNAs. (e) Proportion of antisense lncRNAs and lincRNAs within total lncRNAs. (f) Number of lncRNA-mRNA co-location pairs in the Ginkgo embryos.
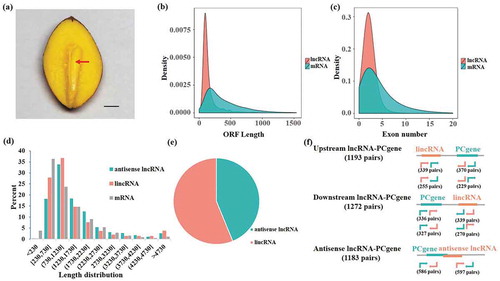
To more clearly characterize the lncRNAs in G. biloba embryos, we compared lncRNAs and mRNAs. Approximately 90% of lncRNA transcripts with open reading frames (ORFs) shorter than 300 bp were present in significantly higher proportions than mRNAs (). The highest exon number of both lncRNAs and mRNAs was two, and lncRNAs tended to have fewer exons than mRNAs on average (). lincRNAs and antisense lncRNAs both had the highest transcript length proportions, ranging from 730 bp to 1730 bp (). Additionally, the co-locations of target genes of the lncRNAs were predicted. Among them, there were 1193 pairs with lncRNA located upstream of the mRNA, 1272 pairs with lncRNA located downstream of the mRNA, and 1183 pairs with lncRNA located on the antisense strand from the mRNA ().
Expression of lncRNAs and mRNAs in different developmental stages of the Ginkgo embryo
Differentially expressed lncRNAs were selected based on statistical significance (q-value < 0.05). A total of 657 differentially expressed lncRNAs were identified in the three developmental stages of the G. biloba embryos. Hierarchical clustering heatmaps of differentially expressed lncRNAs () and stage-specifically expressed lncRNAs (, Supplementary Table S2) revealed significant variations in expression. Specifically, the FPKM distribution of lincRNAs, antisense lncRNAs, and mRNAs of the three development stages (S, N, and F) of G. biloba embryos displayed that lincRNAs had a lower proportion than antisense lncRNAs and mRNAs when the FPKM was greater than 75 ().
Figure 2. Expression of lncRNAs and mRNAs in early (S), middle (N), and late (F) stages of G. biloba embryo development. (a) Cluster heatmap of differentially expressed lncRNAs in embryos during the three stages. (b) Heatmap of specific expression of 30 lncRNAs at three stages of embryo development. (c) Proportion of FPKM distribution of lincRNAs, antisense lncRNAs, and mRNAs in the three stages of embryo development. (d) Number of upregulated or downregulated lncRNAs in N vs. S, F vs. N, and F vs. S. (e) Number of common/specific up- or downregulated lncRNAs in N vs. S, F vs. N, and F vs. S.
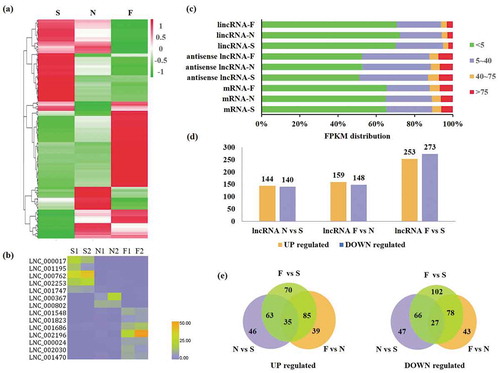
Differences in both up- and downregulated lncRNAs were identified by pairwise comparisons of expression levels among the three different stages. Compared with the early stage, 284 lncRNAs were differentially expressed in the middle stage, including 144 up- and 140 downregulated lncRNAs (). Similarly, 307 lncRNAs were differentially expressed in the mature stage compared to the middle stage, with 159 up- and 148 downregulated lncRNAs (). Lastly, 526 lncRNAs were differentially expressed in the mature stage compared to the early stage, with 253 up- and 273 downregulated lncRNAs (). RNA-seq data was validated by qRT-PCR using six selected lncRNAs differentially expressed over different stages (Supplementary Figure S1, supplementary Table S3)
Further analysis demonstrated that of the total 338 upregulated lncRNAs, 46, 39, and 70 were uniquely upregulated in the N vs. S, F vs. N, and F vs. S comparisons, respectively. Additionally, 35 lncRNAs were upregulated in all three stages (). Of the total 363 downregulated lncRNAs, 47, 43, and 102 were uniquely downregulated in the N vs. S, F vs. N, and F vs. S comparisons, respectively, and 27 lncRNAs were downregulated in all three stages ().
Functional analysis of the differentially expressed lncRNAs
To identify regulatory interactions between lncRNAs and their potential PCgene targets, co-expression analysis of lncRNAs and PCgenes, based on Pearson’s correlation coefficient (PCC), was carried out (Supplementary Table S4). To further investigate the potential function of these differentially expressed lncRNAs in embryos, Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) enrichment analyses were performed and the significantly enriched KEGG pathways (Supplementary Figure 2a) and GO terms (Supplementary Figure 2b, Supplementary Table S5) were analyzed. Among the enriched GO terms, “oxidation-reduction process” and “ubiquitin-protein transferase activity” were significantly enriched in the N vs. S and F vs. S comparisons, respectively (Supplementary Table S5). Analysis of the motifs enriched in lncRNAs associated with these processes is shown in Supplementary Figure 3.
Among the enriched KEGG pathways, “Photosynthesis – antenna proteins”, “Nitrogen metabolism”, and “Plant hormone signal transduction” were the most significantly upregulated pathways in the N vs. S, F vs. N, and F vs. S comparisons, respectively. Meanwhile, “Circadian rhythm – plant”, “Flavonoid biosynthesis”, and “DNA replication” were the most significantly downregulated pathways in the N vs. S, F vs. N, and F vs. S comparisons, respectively. It was also observed that some plant hormone biosynthesis related pathways were significantly enriched (p-value < .05), including “Zeatin biosynthesis”, “Diterpenoid biosynthesis”, and “Brassinosteroid biosynthesis” (Supplementary Figure 2a).
Apart from being candidates for trans-acting pairs, some lncRNA and PCgene pairs may also act as cis-regulators by influencing the expression of adjacent PCgenes. Therefore, we identified and annotated the neighboring genes (within 100 kb) of the lncRNAs. There were a total of 2613 PCgenes co-located with 1804 lncRNAs (Supplementary Table S6). Further interrogation of PCCs of the co-located mRNA-lncRNA pairs was used to observe the correlation with their expression. In total, there were 1491 (40.76%) pairs with no PCC values because of the expression of mRNAs in pairs were not detected in this study. What is more, 235 (6.42%) pairs were of significant positive correlation (R > 0.95) and 8 (0.21%) pairs were of significant negative correlation (R < −0.95). In addition, there were 924 (25.26%) pairs with R between 0.3 and 0.95, and 417 (11.4%) pairs with R between −0.95 and −0.3. These results imply that majority of cis-acting lncRNAs are co-expressed with their targets (Supplementary Tables S6 and S7). These data also suggest that 70 lncRNAs may be involved in the regulation of catalytic activity, according to the GO terms of the target genes, with most of them positively correlated with their target genes and located antisense of their target genes (Supplementary Table S7).
lncRNAs related to plant hormone biosynthesis and signal transduction and circadian rhythm
Endogenous factors, such as plant hormones, have been shown to be associated with embryo development and seed germination.Citation10 To investigate the relationship between lncRNAs and endogenous hormone-related genes in the embryo of G. biloba, we constructed co-expression and co-location networks of lncRNAs and PCgenes involved in plant hormones and found that 50 and 33 lncRNAs might be related to plant hormone signal transduction and plant hormone biosynthesis, respectively (). The expression of genes and lncRNAs related to gibberellin, abscisic acid, auxin, cytokinin, and ethylene signal transduction are displayed in heatmaps (). Most of these genes were significantly upregulated in the late stage in contrast to the early and middle stages; e.g., PP2C (Gb_03279, Gb_13657, Gb_33391, and Gb_27731), SnRK (Gb_02396, Gb_09505, and Gb_32504), and ABF (Gb_16168, Gb_38174, and Gb_40094) in the ABA signal transduction pathway. Fewer genes, such as RGL2 (Gb_20415 and Gb_22850), SLY1 (Gb_08961), and AHP1 (Gb_35949), were significantly downregulated in the late stage compared to the early and middle stages ().
Figure 3. lncRNAs related to plant hormone biosynthesis and signal transduction during Ginkgo embryo development. (a) Co-expression network of lncRNAs and protein coding genes (PCgenes) involved in plant hormone biosynthesis and signal transduction. Circles with red labels represent PCgenes involved in plant hormone signal transduction and circles with black labels represent PCgenes involved in plant hormone biosynthesis. Diamonds represent lncRNAs. Nodes of lncRNAs with one degree were omitted. (b) Co-location network of lncRNAs and PCgenes involved in plant hormone signal transduction. Parallel lines represent lncRNAs located on the antisense strand of the PCgenes. Lines with arrows represent lncRNAs that were located downstream of the PCgenes. (c) Expression of selected lncRNAs and their predicted co-expression target PCgenes involved in plant hormone signal transduction.
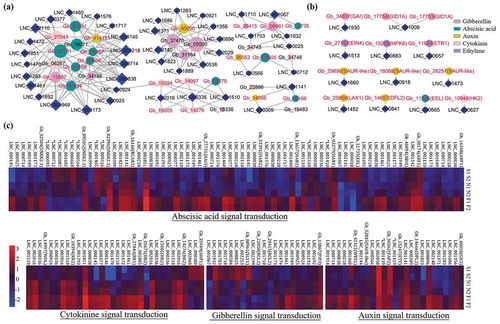
Previous studies have demonstrated that clock genes, such as LHY, CCA, TOC, GI, and PRR7 in circadian rhythm pathways can regulate seed dormancy by influencing plant hormone balance in Arabidopsis.Citation11-Citation13 Therefore, differentially expressed lncRNAs that were significantly co-expressed with genes involved in circadian rhythm were identified (Supplementary Fig. S4). The expression levels of most circadian clock genes, such as LHY (Gb_28874, Gb_25658, and Gb_06934), ZTL (Gb_06336), and PRR7 (Gb_23159 and Gb_06535), was relatively lower in the early embryo development stage but increased in the mature stage (Supplementary Figure S4a). Furthermore, a co-expression network of lncRNAs and PCgenes involved in circadian rhythm was constructed, in which the expression levels of 72 lncRNAs were positively correlated and 10 were negatively correlated with their target genes (Supplementary Figure S4b).
lncRNA transcripts involved in the competing endogenous RNA (ceRNA) regulation network
As a type of ceRNA,Citation3,Citation14 lncRNAs can compete with PCgenes in binding miRNAs. To search target lncRNAs of miRNAs, lncRNAs that may be precursors of miRNAs should be filtered out first according to the homology of lncRNAs and miRNA precursors, and the same expression trend of lncRNAs (average FPKM of each lncRNA was over three in each stage) and mature miRNAs during three different stages. A total of 15 lncRNAs as miRNA precursors of G. biloba were predicted (Supplementary Table S8) and an example of the secondary structure of a lncRNA predicted as miRNA precursor is depicted in .
Figure 4. Analysis of lncRNAs involved in ceRNA interaction networks. (a) Example of lncRNAs predicted as miRNA precursors. (b) Example of lncRNAs predicted as miRNA target mimics. (c) Interaction network of lncRNA-miRNA-mRNA. Triangles represent miRNAs, circles represent lncRNAs, and squares represent PCgenes. (d,e) Quantification of lncRNA, miRNA and mRNA within two up-down-up ceRNA interactions in the F vs S comparison by RNA-Seq.
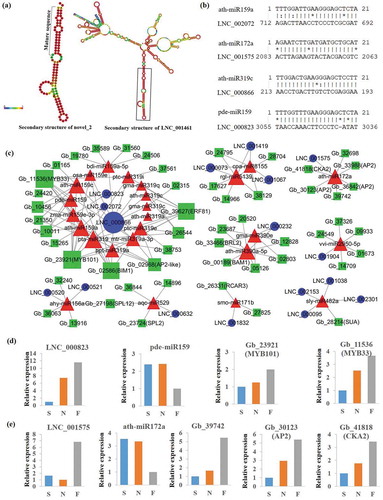
psRobot was used to predict sequence-based targeted relationships between miRNA and lncRNA and miRNA and gene pairs. Examples of targeted relationships between miRNAs and lncRNAs are displayed in . Finally, a lncRNA (17)-miRNA (25)-PCgene (52) network was constructed for the G. biloba embryo (). According to the GO enrichment result of 52 PCgenes in the network, we found that there were eight genes related to transcription factor activity, twelve genes related to protein kinase activity, and three genes related to chromatin modification (Supplementary Table S9). This imply that lncRNAs in this network may play important roles through acting as ceRNA of genes related to transcription factor activity, protein kinase activity and chromatin modification.
In this network, 11 miRNA families members were covered, among which there were several miRNAs associated with seed development, such as miR390,Citation15 or seed dormancy and germinate, such as miR159,Citation16 miR156, and miR172.Citation17-Citation19 MYB33 and MYB101 were both targets of miR159, and one study showed that MYB33 and MYB101 functioned as positive regulators of ABA responses in germinating Arabidopsis thaliana seeds and ABA-induced accumulation of miR159 directed MYB33 and MYB101 transcript degradation to desensitize hormone signaling.Citation16 In this study, we find three lncRNAs (lnc000823, lnc002072, lnc000866) were predicted as target mimics of miR159, which targeted Gb_11536 (MYB33) and Gb_23921 (MYB101), indicating that this three lncRNAs may be involved in ABA signal regulation during Ginkgo seed maturation (). In addition, lnc000520, lnc000521 were predicted as target mimics of any-miR156a that targeted Gb_27198 (SPL12), and lnc001575 was predicted as target mimics of ath-miR172a to protect three AP2 genes (Gb_33988, Gb_36842, Gb_30123) from degradation. lnc001832 were predicted as ceRNA of Gb_26331 (RCAR3; regulatory component of ABA receptor 3). lnc001038, lnc002301, lnc000095 and lnc002153 were putative ceRNAs of Gb_28214 (SUA; splicing factor suppressor of ABI3-5), which were involved in ABA signal transduction. These results suggest the potential important roles of lncRNAs involved in these networks during seed development.
Based on the “ceRNA hypothesis”, mRNAs and lncRNAs in ceRNA interactions are required to have positively correlated expression, and both of them are required to have negatively correlated expression with their miRNA regulators, as miRNAs are mostly known to repress the expression of their targets.Citation20 Therefore, we further employed expression-based filtering conditions to identify stage-specific ceRNA interactions. Differently expressed (q-value < 0.05) lncRNAs, miRNAs and mRNAs that involved in lncRNA(17)-miRNA(25)-PCgene(52) network were screened out. Then, lncRNA-miRNA-mRNA network with up-down-up or down-up-down expression pattern in pairwise comparisons of three development stages was shown in Supplementary Table S10. For example, as shown in , both two MYB transcripts and lnc000823 were up-regulated in the F vs S comparison, and all of them were negatively correlated with pde-miR159 which was down-regulated in the F vs S comparison. Similarly, expression pattern of lnc001575- ath-miR172a- Gb_30123/Gb_39742/Gb_41818 network was up-down-up in the F vs S comparison (). These expression data further enhanced the significance of putative ceRNA interactions.
Discussion
There is increasing evidence that the abundance and diversity of lncRNAs fulfill a wide variety of regulatory roles in plant growth and development via versatile mechanisms.Citation1,Citation2 To understand the roles of lncRNAs in embryo development of Gingko biloba, we explored lncRNAs within G. biloba embryos. Here, we have identified 1307 lincRNAs and 1019 antisense lncRNAs in G. biloba embryos, which is far more than those found in G. biloba leaves (762 lincRNAs and 211 antisense lncRNAs).Citation21 This abundance is likely due to the active differentiation and developmental processes of embryos. Additionally, the identified lncRNAs from G. biloba embryos shared most of the common features of lncRNAs compared with PCgenes reported in other plants, such as lower exon numbers, lower expression, and shorter ORF length,Citation22 supporting the theory that lncRNAs share a common and ancient evolutionary origin and have undergone rapid sequence evolution.Citation23
lncRNAs have been identified in the seeds of many plants, such as maizeCitation4 and rice,Citation6 and have been shown to be related to seed development. Here, functional annotation of target genes of lncRNAs demonstrated that many substance biosynthetic and metabolic processes, including those of flavonoid, fatty acid, and glutathione mainly focused on the middle stage in embryo development of G. biloba, suggesting the potential involvement of lncRNAs in the metabolism of primary and secondary substances during the middle stage of embryo development. Many target genes of lncRNAs connected with transcriptional regulation, circadian rhythm, and plant hormone signal transduction were highly expressed in the mature stage, indicating that lncRNAs actively participate in embryo maturation and seed germination. Networks of lncRNAs and PCgenes identified lncRNAs with potential roles in plant hormone biosynthesis (33 lncRNAs), signal transduction (50 lncRNAs), and circadian rhythm (76 lncRNAs). Additionally, some lncRNAs might function as ceRNAs, acting as regulators of chromatin modification or transcription factor activity. Consistent with the complexity and diversity of lncRNA modes of action, there are many possible ways in which G. biloba lncRNAs regulated embryo development and dormancy release.
Previous studies have indicated that lncRNAs may be part of a broad epigenetic regulatory network.Citation24 Many lncRNAs bind to chromatin-modifying proteins and recruit their catalytic activity to specific sites within the genome, thereby modulating chromatin states and impacting gene expression.Citation25 Especially during early seed development, chromatin modification and remodeling directly dictate the permissive or repressive transcriptional state of a gene, or a whole genome, to orchestrate embryo-specific gene expression programs.Citation26 From the present study, we found three ceRNA network-associated PCgenes (Gb_23921, Gb_11536, Gb_09933) were related to chromatin modification based on the function annotation (Supplementary Table S9). Four lncRNAs (lnc000866, lnc000823, lnc002072 and lnc001904) connected with these three mRNAs through eleven miRNAs belonging to three miRNA families (miR159, miR319, and miR2950), implying that these four lncRNAs might contribute to embryo development through chromatin modifications. With the emergence of genome-wide chromatin modification profiling, further studies are required to elucidate these specific mechanisms, such as genome-wide comparison of DNA or histone modifications between specific lncRNA knockout and overexpression in embryos.
Normal circadian clock gene function is essential for the response to dormancy-breaking signals, such as plant hormones in seeds.Citation11 Consistent with the role of clock genes in seed dormancy control, LHY, PRR7, and GI expression was transcriptionally induced during the late embryo development stage, implying the transition from dormancy to germination. Although lncRNAs have been previously linked to circadian regulation in mouse livers,Citation27 little is known about lncRNAs related to circadian rhythm in plants. This study identified a set of lncRNAs related to circadian rhythms in Ginkgo embryos that might have regulatory roles in Ginkgo embryo development and dormancy. Further experiments are required to understand the precise mechanism of lncRNAs affecting and responding to embryo development and dormancy.
Materials and methods
Description of RNA-seq datasets
A transcriptome dataset of G. biloba embryos at different stages of development was analyzed to identify lncRNAs. The dataset included two biological replicates of six samples at three developmental stages (Supplementary Table S11): early [0 day post harvest and sand-storage on September 2015 (S)], middle [70 days post sand-storage on November 2015 (N)], and late [150 days post sand-storage on February 2016 (F)].The libraries were sent to Novogene for transcriptome sequencing on an Illumina HiSeq 4000 platform, which produced paired-end 150 bp reads. The dataset was deposited in the Sequence Read Archive under the accession number SRP189045.
Bioinformatics identification of Ginkgo embryo lncRNAs
Quality-controlled reads were obtained by discarding adapter sequences, contaminating sequences, and low quality reads from the raw data. The quality-controlled reads were aligned to the Ginkgo reference genome (http://gigadb.org/dataset/100209) using the TopHat 2.0 program. After alignment, Cufflinks was used to assemble the transcripts of each sample and all of the assemblies were merged together to form a consensus transcriptome assembly using “Cuffmerge” program in the Cufflinks software package. The expression level (FPKM value) of each transcript was calculated with Cuffdiff software.
Transcripts were subjected to the following pipeline to identify high-confidence lncRNAs. Briefly, transcripts with sequence length of less than 200 nucleotides and FPKM less than 0.5 were removed. The remaining transcripts were compared with the Ginkgo reference genome annotations using Cuffcompare, and transcripts that overlapped with known genes in the sense strand were discarded. Finally, the CPC and CNCI software packages were used to filter transcripts with coding potential, and those with a significant coding potential (CPC score ≥ 0 or CNCI score ≥ 0) were filtered out. The remaining transcripts were considered to be reliable lncRNAs. The identified lncRNAs were further divided into two classes, long intergenic noncoding RNAs (lincRNAs) and antisense lncRNAs using the Cuffcompare program in the Cufflinks suite. The references of bioinformatics software and databases used are summarized in Supplementary Table S12.
Differential expression analysis
Cuffdiff software was employed to identify differentially expressed lncRNAs between pairs of samples at the three stages (q-value < 0.05). Hierarchical clustering heatmaps were generated with the Heml v1.0 and OmicShare tools. lncRNAs expressed at specific stages were selected with expression restriction (FPKM > 5 in one stage and FPKM < 1 in all the others both in two biological replicates, p-value < .05). The references of bioinformatics software and databases used are summarized in Supplementary Table S12.
Function prediction of the differentially expressed lncRNAs
The identified lncRNAs were annotated with functional annotation of lncRNA co-expression and co-location targets. For co-expression analysis, PCCs between the FPKM values of mRNAs and lncRNAs were calculated, and the cutoff PCC value (> 0.99 or < −0.99) was used to screen the putative co-expression target mRNA. For co-location analysis, putative co-location target mRNA was searched from 100 kb upstream and downstream of the lncRNA. To identify potential biological functions of the significant differentially expressed lncRNAs (|foldchange| ≥ 2 and q-value < 0.05) during the development and dormancy of Ginkgo embryos, the co-expressed (|foldchange| ≥ 2 and q-value < 0.05) and co-located target mRNAs were subjected to KEGG and GO enrichment analysis separately. Finally, the co-expression and co-location networks were visualized using Cytoscape (v3.5.0) software. MEME was used to discover motifs (widths = 6–50, allowing zero or one motif per sequence, E-value < 0.05) enriched in lncRNAs associated with specific functions. The references of bioinformatics software and databases used are summarized in Supplementary Table S12.
Construction and analysis of the lncRNA-miRNA-mRNA network
The G. biloba embryo miRNA dataset during embryo development (accession numbers: SRP189045) was used to predict the target relationships between miRNA and RNAs (mRNAs and lncRNAs) with psRobot. Based on targeted relationship between lncRNAs, mRNAs, and miRNAs, a lncRNA-miRNA-PCgene network was constructed using Cytoscape software (v3.5.0). The references of bioinformatics software and databases used are summarized in Supplementary Table S12.
Disclosure of potential conflicts of interest
The authors declare no conflict of interest.
Data archiving statement
Raw data have been deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra/) under Bioproject accession PRJNA528417.
Supplemental Material
Download MS Excel (15.5 MB)Supplemental Material
Download MS Word (2.8 MB)Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
Related Research Data
References
- Xu W, Yang T, Wang B, Han B, Zhou H, Wang Y, Li D, Liu A. Differential expression networks and inheritance patterns of long non-coding RNAs in castor bean seeds. Plant J. 2018;95:1–8. doi:10.1111/tpj.13953.
- Bouckenheimer J, Assou S, Riquier S, Hou C, Philippe N, Sansac C, Lavabre-Bertrand T, Commes T, Lemaître JM, Boureux A, et al. Long non-coding RNAs in human early embryonic development and their potential in ART. Hum Reprod Update. 2016;23:19–40. doi:10.1093/humupd/dmw035.
- Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505:344–352. doi:10.1038/nature12986.
- Zhu M, Zhang M, Xing L, Li W, Jiang H, Wang L, Xu M. Transcriptomic analysis of long non-coding RNAs and coding genes uncovers a complex regulatory network that is involved in maize seed development. Genes (Basel). 2017:8. doi:10.3390/genes8100274.
- Zhang YC, Liao JY, Li ZY, Yu Y, Zhang JP, Li QF, Qu LH, Shu WS, Chen YQ. Genome-wide screening and functional analysis identify a large number of long noncoding RNAs involved in the sexual reproduction of rice. Genome Biol. 2014;15:512. doi:10.1186/s13059-014-0512-1.
- Kiegle EA, Garden A, Lacchini E, Kater MM. A genomic view of alternative splicing of long non-coding rnas during rice seed development reveals extensive splicing and lncRNA gene families. Front Plant Sci. 2018:9. doi:10.3389/fpls.2018.00115.
- Shen E, Zhu X, Hua S, Chen H, Ye C, Zhou L, Liu Q, Zhu Q, Fan L, Chen X. Genome-wide identification of oil biosynthesis-related long non-coding RNAs in allopolyploid Brassica napus. BMC Genomics. 2018;19:1–13. doi:10.1186/s12864-018-5117-8.
- Yin DD, Li SS, Shu QY, Gu ZY, Wu Q, Feng CY, Xu WZ, Wang LS. Identification of microRNAs and long non-coding RNAs involved in fatty acid biosynthesis in tree peony seeds. Gene. 2018;666:72–82. doi:10.1016/j.gene.2018.05.011.
- Baskin JM, Baskin CC. A classification system for seed dormancy. Seed Sci Res. 2004;14:1–16. doi:10.1079/SSR2003150.
- Liu Y, Fang J, Xu F, Chu C, Schläppi MR, Wang Y, Chu C. Expression patterns of ABA and GA metabolism genes and hormone levels during rice seed development and imbibition: A comparison of dormant and non-dormant rice cultivars. J Genet Genomics. 2014;41:327–338. doi:10.1016/j.jgg.2014.04.004.
- Penfield S, Hall A. A role for multiple circadian clock genes in the response to signals that break seed dormancy in Arabidopsis. Plant Cell Online. 2009;21:1722–1732. doi:10.1105/tpc.108.064022.
- Footitt S, Ölçer-Footitt H, Hambidge AJ, Finch-Savage WE. A laboratory simulation of Arabidopsis seed dormancy cycling provides new insight into its regulation by clock genes and the dormancy-related genes DOG1, MFT, CIPK23 and PHYA. Plant Cell Environ. 2017;40:1474–1486. doi:10.1111/pce.12940.
- Adams S, Grundy J, Veflingstad SR, Dyer NP, Hannah MA, Ott S, Carré IA. Circadian control of abscisic acid biosynthesis and signalling pathways revealed by genome-wide analysis of LHY binding targets. New Phytol. 2018;220:893–907. doi:10.1111/nph.15415.
- Liu S, Wu L, Qi H, Xu M. LncRNA/circRNA–miRNA–mRNA networks regulate the development of root and shoot meristems of populus. Ind Crops Prod. 2019;133:333–347. doi:10.1016/j.indcrop.2019.03.048.
- Zhao YT, Wang M, Fu SX, Yang WC, Qi CK, Wang XJ. Small RNA profiling in two Brassica napus cultivars identifies microRNAs with oil production-and development-correlated expression and new small RNA classes. Plant Physiol. 2012;158(2):813–823. doi:10.1104/pp.111.187666.
- Reyes JL, Chua NH. ABA induction of miR159 controls transcript levels of two MYB factors during Arabidopsis seed germination. Plant J. 2007;49:592–606. doi:10.1111/j.1365-313X.2006.02980.x.
- Nonogaki H. microRNA gene regulation cascades during early stages of plant development. Plant Cell Physiol. 2010;51(11):1840–1846. doi:10.1093/pcp/pcq154.
- Das SS, Karmakar P, Nandi AK, Sanan-Mishra N. Small RNA mediated egulation of seed germination. Front Plant Sci. 2015;6:828. doi:10.3389/fpls.2015.00828.
- Nodine MD, Bartel DP. MicroRNAs prevent precocious gene expression and enable pattern formation during plant embryogenesis. Genes Dev. 2010;24(23):2678–2692. doi:10.1101/gad.1986710.
- Zhou X, Liu J, Wang W. Construction and investigation of breast-cancer-specific ceRNA network based on the mRNA and miRNA expression data. IET Syst Biol. 2014;8:96–103. doi:10.1049/iet-syb.2013.0025.
- Wang L, Xia X, Jiang H, Lu Z, Cui J, Cao F, Jin B. Genome-wide identification and characterization of novel lncRNAs in Ginkgo biloba. Trees - Struct Funct. 2018;32:1429–1442. doi:10.1007/s00468-018-1724-x.
- Liu Y, Li M, Bo X, Li T, Ma L, Zhai T, Huang T. Systematic analysis of long non-coding RNAs and mRNAs in the ovaries of duroc pigs during different follicular stages using RNA sequencing. Int J Mol Sci. 2018:19. doi:10.3390/ijms19061722.
- Ulitsky I, Shkumatava A, Jan CH, Sive H, Bartel DP. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell. 2011;147:1537–1550. doi:10.1016/j.cell.2011.11.055.
- Ghosh S, Sati S, Sengupta S, Scaria V. Distinct patterns of epigenetic marks and transcription factor binding sites across promoters of sense-intronic long noncoding RNAs. J Genet. 2015;94:17–25. doi:10.1007/s12041-015-0484-2.
- Mercer TR, Mattick JS. Structure and function of long noncoding RNAs in epigenetic regulation. Nat Struct Mol Biol. 2013;20:300–307. doi:10.1038/nsmb.2480.
- Baroux C, Pien S, Grossniklaus U. Chromatin modification and remodeling during early seed development. Curr Opin Genet Dev. 2007;17:473–479. doi:10.1016/j.gde.2007.09.004.
- Fan Z, Zhao M, Joshi PD, Li P, Zhang Y, Guo W, Xu Y, Wang H, Zhao Z, Yan J. A class of circadian long non-coding RNAs mark enhancers modulating long-range circadian gene regulation. Nucleic Acids Res. 2017;45:5720–5738. doi:10.1093/nar/gkx156.