
ABSTRACT
Selaginella tamariscina is a lycophyta species that survives under extremely dry conditions via the mechanism of resurrection. This phenomenon involves the regulation of numerous genes that play vital roles in desiccation tolerance and subsequent rehydration. To identify resurrection-related genes, we analyzed the transcriptome between dehydration conditions and rehydration conditions of S. tamariscina. The de novo assembly generated 124,417 transcripts with an average size of 1,000 bp and 87,754 unigenes. Among these genes, 1,267 genes and 634 genes were up and down regulated by rehydration compared to dehydration. To understand gene function, we annotated Gene Ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG). The unigenes encoding early light-inducible protein (ELIP) were down-regulated, whereas pentatricopeptide repeat-containing protein (PPR), late embryogenesis abundant proteins (LEA), sucrose nonfermenting protein (SNF), trehalose phosphate phosphatase (TPP), trehalose phosphate synthase (TPS), and ABC transporter G family (ABCG) were significantly up-regulated in response to rehydration conditions by differentially expressed genes (DEGs) analysis. Several studies provide evidence that these genes play a role in stress environment. The ELIP and PPR genes are involved in chloroplast protection during dehydration and rehydration. LEA, SNF, and trehalose genes are known to be oxidant scavengers that protect the cell structure from the deleterious effect of drought. TPP and TPS genes were found in the starch and sucrose metabolism pathways, which are essential sugar-signaling metabolites regulating plant metabolism and other biological processes. ABC-G gene interacts with abscisic acid (ABA) phytohormone in the stomata opening during stress conditions. Our findings provide valuable information and candidate resurrection genes for future functional analysis aimed at improving the drought tolerance of crop plants.
Introduction
Selaginella tamariscina is a primitive resurrection plant that has the ability to resist extreme dehydration conditions and can be retained to a normal form upon rehydration.Citation1 The phenomenon of resurrection has been mostly studied in cyanobacteria and plants.Citation2,Citation3 Among these species, more than 300 species of angiosperms have been known as resurrection plants.Citation4,Citation5 Most Salaginella species have the ability to survive under severe water stress, with almost full loss of protoplasmic water.Citation6 Resurrection plants undergo several physiological and metabolic mechanisms to sustain desiccation.Citation7 Plant modifications such as curling and folding confer drought tolerance by limiting light and forming reactive oxygen species (ROS).Citation8,Citation9 During dehydration, the photosynthetic apparatus is damaged;Citation10 therefore, resurrection plants consist of inducible repair mechanisms that maintain their photosynthetic apparatus.Citation11,Citation12
Abscisic acid (ABA) plays an important role during water deficit conditions.Citation13 Several ABA response genes have been discovered to date. ABA plays a central role in various stress conditions through network signaling with several gene families.Citation14 Plants have a mechanism to adapt under light stress through the mechanism attributed to the chlorophylla/b-binding protein (CAB) family to protect chloroplasts.Citation15 Therefore, early light inducible proteins (ELIPs) protect plant leaves during light stress and play a major role in photoprotection.Citation16 During dehydration, the rate of chlorophyll synthesis and photosynthesis is reduced;Citation17 therefore, a large number of pentatricopeptide repeated proteins(PPRs) are required for chloroplast development.Citation18 When plants undergo drought stress, energy-generating organelle mitochondria are supposed to be damaged.Citation19 In order to protect, LEA proteins and make biochemical and secondary structures to withstand in desiccation stress.Citation20 Recently, trehalose biosynthesis pathway genes have been studied that respond to drought.Citation21 These trehalose-related genes are responsible for inhibiting sucrose non-fermenting (SNF) proteins to regulate the energy during stress conditions.Citation22 SNF proteins form an interaction network with ABA and function during abiotic stress.Citation23 During desiccation, ABC transporters regulate hormones and secondary metabolites.Citation24 Additionally, ABCG gene families have been identified in the moss Physcomitrella patens for adaptation to extreme environmental conditions.Citation24
Resurrection plants are studied to understand and identify the genes related to the mechanism of desiccation tolerance.Citation10 To elucidate the mechanism of desiccation, several genomic approaches have been discovered.Citation25 In our study, to identify the genes involved in resurrection, we analyzed differentially expressed genes (DEGs) based on comparison of the transcriptome among dehydrated and rehydrated leaves of S. tamariscina. Since angiosperm resurrection plants have been extensively studied, we characterized the major genes and their functions involved in desiccation. For this knowledge, the identification of potential candidate genes associated with resurrection based on transcriptome and DEG analyses will improve our understanding of the regulation and function of the gene response to dehydration and rehydration.
Materials and methods
Plant materials and sample preparation for transcriptome analysis
Selaginella tamariscina plants were grown in pots under a controlled environment plant growth room. The plants were divided into two groups, with a plastic film as a barrier for dehydration and rehydration. The plants were grown with regular watering prior to desiccation treatment. For the desiccation experiment, 2 months old grown plants with similar-sized aerial parts were selected. Water was withheld for 7 days, and the morphology of the plants was observed. After 7 days only one group was watered regularly with bottle spray until the leaves fully expanded, while the other was left water-deprived. After complete rehydration, leaf tissues were harvested from each group of plants and placed immediately in liquid nitrogen for total RNA isolation.
Water content and phenotype observation
To evaluate the resurrection phenomenon, the leaves were taken from the separately grown rehydrated pots. The leaves (approximately 3–5 g) were plugged from the pot and left on a dry laboratory bench. Different relative water content (RWC) was provided. The fresh leaves were subjected RWC of 70% and slightly dropped to 30%. The plant morphology was observed with minimum RWC. Then for rehydration, 30% RWC leaves were placed on water-soaked facial tissue in a petri dish and covered with lids. The tissues were sprayed with water every 2 hours and 4 hours. The excess water was removed from the surface by blotting with fresh facial tissues. Finally, the morphology of the rehydrated plants was observed.
RNA extraction and sequencing
Total ribonucleic acid (RNA) was extracted from leaf tissues using the RNeasy Plant Mini Kit (Cat No./ID: 74904, Qiagen, USA) according to the manufacturer’s instructions. The quality and concentration of the RNA were assessed using an Agilent Bioanalyzer (Agilent Technology, USA) and a Nanodrop spectrophotometer (Thermo Fisher Scientific, USA) with the following parameters: RNA integrity number (RIN) ≥ 7, 28S:18S> 1, and ratio of optical density at 260 and 280 nm (OD260/280) ≥ 2. RNA-Sequencing was performed using the mRNA isolated from the dehydrated and rehydrated plants leaf, with three biological replicates. cDNA libraries were made using a TruSeq Stranded mRNA kit (Cat. No. RS-122–2101, Illumina, USA). The quality of the sample libraries was assessed using the Agilent Bioanalyzer 2100 system. The libraries were processed for high-throughput DNA sequencing on an IlluminaNextSeq 2000 with the 150 bp paired-end (PE) method.
Transcriptome assembly, annotation, and functional analysis
The next-generation sequencing (NGS) reads were filtered by discarding reads with >20% N bases, Phred quality ≤ Q20 and length > 50 bp. De novo assembly was performed using Trinity (https://github.com/trinityrnaseq)Citation26 and CD-HIT (http://weizhongli-lab.org/cd-hit/).Citation27 Transcriptomes were searched for matching sequences in the National Center for Biotechnology Information (NCBI) database using NCBI BLAST, and coding sequence prediction and function prediction were performed with InterProScan (https://www.ebi.ac.uk/interpro)Citation28 and TCC (https://github.com/TransDecoder/TransDecoder) programs. Transcriptome quantification and differentially expressed gene (DEG) analyses were carried out using the software applications RSEM (https://github.com/deweylab/RSEM)Citation29 and TCC (https://bioconductor.org/packages/release/bioc/html/TCC.html),Citation30 respectively. Gene Ontology (GO) enrichment analysis was performed with GO-seq (https://bioconductor.org/packages/release/bioc/html/goseq.html),Citation31 with a p-value criterion of < 0.001 for the categories of “molecular function”, “biological process” and “cellular component”. The KEGG pathway was analyzed by the KEGG database (http://www.genome.jp/kegg/pathway.html).
Screening of differentially expressed genes (DEGs)
Expression level analysis was carried out with the filtered high-quality raw reads by counting mapped reads in the unigene set using RSEM softwareCitation29 and TCC.Citation30 The expression value for each gene was calculated with the fragments per kilobase of transcript per million mapped reads (FPKM) method. The significant DEGs were confirmed by Fisher’s exact test (p ≤ 0.05). Additionally, the p-value was adjusted for multiple comparisons by calculating the false discovery rate (FDR) upto 5%; this Q-value was used to assess differences using multiple test adjustments. Visualization analysis of the volcano plot and heat map clustering of the DEGs were performed by an in-house R script. Finally, the gene response to resurrection was analyzed and selected for further functional analysis.
Results
Resurrection phenomenon of S. tamariscina
Two experiments were conducted, one for transcriptome analysis, another for water content phenotype observation. First, the aerial lycophyll leaves of S. tamariscina were observed. The water was withheld for 7 days, and the plants were found to be almost dried under dehydration conditions, whereas those irrigated with bottle spray were fully recovered (). Second, the resurrection phenomenon was observed in some lycophyll. After water deprivation with different RWC, the plant curled up and changed its appearance. The relative RWC slightly dropped to 70% and severely to 30%, which formed the aerial part into a ball shape. When water was provided again, the aerial part was fully recovered with the highest water content (Supplementary ). During dehydration and rehydration, we observed morphological changes in the aerial part of the plant, which showed the complete phenomenon of resurrection.
Figure 1. Morphology of aerial lycophyll of S. tamariscina exhibiting resurrection phenomenon upon dehydration and rehydration. The plants were grown in pots under a controlled environment plant growth room until fully matured. White line represents the plastic barrier, divided into two part (dehydration and rehydration). Both groups of plants were subjected to water stress for a week. Left panel (dehydration) shows the plants with continuous water deprivation and right panel (rehydration) represents the plants irrigated by a bottle spray after desiccation. The dehydrated plants exhibited gradual curling and folding of lycophyll due to water deficit and rehydrated plants show a normal state after being completely watered.

De novo assembly
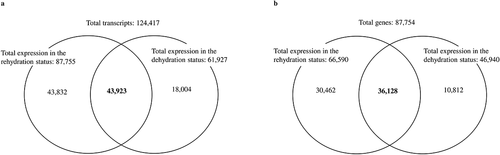
From two different stages of plants under dehydration and rehydration, lycophyll was used for RNA-Seq analysis. Total RNA from three independent replicates was pooled for mRNA and cDNA synthesis and library preparation. There was no reference genome for S. tamariscina, so de novo assembly was selected. The IlluminaNextSeq 2000 150 PE paired sequencing generated 29,233,500 qualified sequence reads with 7,632,894,419 bp length for the rehydration stage and 28,897,467 reads with 7,545,155,886 bp length for the dehydration stage (). Both sets of filtered reads were pooled to construct a transcriptome reference by de novo assembly using Trinity and CD-HIT software. The de novo assembly generated 124,417 transcripts that varied in size from 224 bp to 19,122 bp, with an average size of 1,000 bp. Of the de novo assembled transcripts, 87,754 were revealed to be unigenes through homology searches using NCBI-BLASTx and InterProScan tools (, ). The distribution of the unigenes according to the length obtained is shown in Supplementary . Of those obtained unigenes, 63,740 (72.6%) were matched with plant genes with the highest BLAST scores in the BLAST analysis. The average length of the annotated unigenes was 1,136 bp, with a minimum of 224 bp and a maximum of 19,122bp, and most of the unigenes were less than 1.5 kb in length ().
Table 1. Summary of raw reads and filtered reads of RNA sequencing
Table 2. Summary of contigs and length by de novo assembly
Figure 2. Differentially abundant transcripts and annotated genes upon dehydration and rehydration. a) The Venn diagram shows the expression of total transcripts between rehydration and dehydration leaves obtained from the assembly. b) The statistics expression of total annotated genes between rehydration and dehydration leaves. A total of 124,417 transcripts were obtained from the de novo assembly, among which 87,754 unigenes were annotated through different homology searches.
Analysis of differentially expressed genes (DEGs)
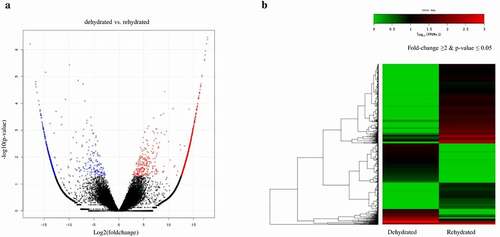
To gain a comprehensive overview of the S. tamariscina transcriptome, a study of differentially expressed genes was conducted. Based on the number of reads mapped onto the reference, the expression level and quantification of each gene were calculated. A total of 124,427 transcripts generated from the sequencing 61,927 were expressed in the dehydrated stage, and 87,755 were expressed in rehydrated stage, among which 43,923 were commonly expressed in both stages (, Supplementary ). From a total of 87,754 annotated genes, 66,590 were expressed in hydrated tissues, and 30,462 were expressed in dehydrated tissues, accounting for 45.7% of the expressed genes in hydrated tissues. Additionally, 46,940 genes were expressed in dehydrated tissues, and 10,812 were expressed only in the dehydrated tissues, which accounted for 23% of the genes expressed in dehydrated tissues (). The number of genes with reduced expression in dehydrated tissues compared to hydrated tissues was 1.67 times greater than the number of genes with increased expression. The number of DEGs was reduced to 84,247 at a fold change threshold of 2. Then, it was further reduced to 1,901, in which 1,267 were up regulatedand 634 were down regulated in the rehydrated state at a p-value ≤ 0.05. Of the 1,901 DEGs, the number with reduced expression in rehydrated tissues was approximately half the number with increased expression (). The expression of DEGs between dehydration and rehydration was visualized with a volcano plot () and heat map with fold change ≥ 2 and p-value ≤ 0.05 (), which shows that most of the genes were regulated in rehydration. Among the total DEGs, we found that the maximum number of unigenes was up regulated during rehydration. This indicates that the maximum number of genes was responsible for the regeneration of the plants after rehydration.
Table 3. Statistical status of differentially expressed genes between dehydrated and rehydrated samples
Figure 3. Expression profiles of differentially expressed genes (DEGs) in dehydrated and rehydrated samples. a) Volcano plot of DEGs, red dots are the genes that are up regulated and blue dots are the down regulated genes in rehydrated samples. The x-axis plots the distribution of fold change and the y-axis plots the logarithm of p-value of each identified DEGs. b) Schematic representation of heat map clustering of DEGs. Green color indicates the lowest expression, black as intermediate expression, and red indicates the highest expression within the DEGs.
Gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) pathway analyses
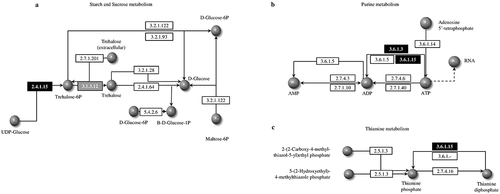
To determine the association of DEGs involved in resurrection, we performed functional GO annotation and KEGG pathway analysis. Using the GO-Seq platform, we classified DEGs of dehydration and hydration into “biological process”, “cellular component” and “molecular function” categories. There was no visual difference between transcripts from hydrated and dehydrated tissues. The numbers of DEGs among the transcripts from hydrated tissues were 94,913 under cellular components, 75,690 under molecular functions, and 45,373 under biological processes. Among the 1,901 DEGs, 30 unigenes were classified under biological processes and 15 under molecular functions, and there were only 7 functional categories under cellular components (). Both up regulated and down regulated DEGs showed almost the same functional classifications. Under biological processes, approximately 60% of the DEGs were in the top four functional categories of “cellular process”, “metabolic process”, “response to stimulus” and “biological regulation”. Moreover, there were only two predominant functional categories each under molecular functions and in cellular components: “catalytic activity” (43%) and “binding” (43%) for the latter and “cellular anatomical entity” (50–52%) and “intracellular” (40–41%). We found that more unigenes were involved in biological processes and that a minimum number of unigenes were involved in cellular components during resurrection. Among the total DEGs, 29 unigenes were annotated to different pathways. Among them, 2 unigenes were annotated to the starch and sucrose metabolism pathway, 18 unigenes were annotated to the purine metabolism pathway, and 9 unigenes were annotated to the thiamine metabolism pathway (). These are the pathway genes related to the resurrection phenomenon in S. tamariscina. The family member genes TPP4 and TPS1 encode trehalose-phosphate phosphatase-4 and alpha-alpha trehalose-phosphate synthase-1, respectively, which are involved in the trehalose synthesis pathway (). KEGG pathway analysis showed the products of ABCG were involved in thiamine metabolism and purine metabolism(, c).
Table 4. KEGG pathway genes related to resurrection phenomenon
Figure 4. The classification of gene ontology (GO) of unigenes with three independent categories: biological process, molecular function, and cellular component. A total of 1,901 DEGs, 30 were annotated to biological, 15 under molecular functions, and 7 under the cellular component. Y-axis indicates the number of unigenes while the x-axis represents the GO category.
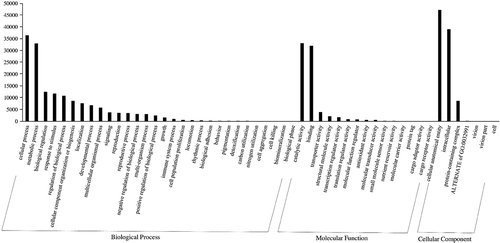
Figure 5. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis. a) Trehalose biosynthesis in starch and sucrose metabolism pathway. This pathway consists of the KEGG enzyme number information of trehalose. The Black highlighted boxes represent trehalose phosphate synthetase (TPS1) and the gray box indicates trehalose phosphate phosphatase (TPP4). These two genes (2.4.1.15 and 3.1.3.12) are responsible for the formation of trehalose necessary for desiccation tolerance. b) Purine metabolism pathway and c) thiamine metabolism pathway involving ABC-G genes that interacts with different enzymes information of phosphate. Phosphate promotes rapid recovery of plant physiological functions when re-watering after drought stress. The number in the boxes are KEGG enzyme and black highlighted boxes represent the ABC-G gene.
Identification of resurrection genes
We analyzed the functional annotations of the DEGs that exhibited the greatest difference in expression in response to dehydration and rehydration. The top unigenes that were significantly expressed are listed in . We shortlisted reported genes that play a significant role in drought tolerance. Early light inducible proteins (ELIPs), pentatricopeptide repeat-containing protein (PENTA/PPR), late embryogenesis abundant proteins (LEAs), sucrose non-fermenting proteins (SNFs), threalose phosphate phosphatase (TPP), trehalose phosphate synthase (TPS), and ABC transporter G family member (ABCG) were abundantly expressed during the resurrection process. Our results showed that a large number of DEGs were enriched during the rehydration process. Most of the identified genes are up regulated by resurrection. The unigenes encoding ELIP found to be down regulated by rehydration, whereas the unigenes encoding PENTA/PPR, LEA, SNF, TPP, TPS andABCG were found up regulated by rehydration. These are the genes reported in previous research under different desiccation conditions with resurrection phenomena. We predict that these genes play a significant role during the rehydration process and keep plants alive under desiccation conditions.
Table 5. Significantly expressed DEGs involved in resurrection phenomenon of S. tamariscina.
Discussion
S. tamariscina species have been extensively studied in relation to desiccation tolerance. Resurrection phenomenon studies in S. tamariscina have revealed the relationship between the morphology and desiccation tolerance mechanism. Therefore, the comparative RNA-Seq analysis in our study included the characterization of the unigenes response to the desiccation tolerance based on previously reported research. We analyzed the DEGs between dehydrated and rehydrated tissues among the DEGs we identified and discussed the expression of the genes involved in desiccation tolerance and subsequent rehydration.
Generation of ROS is amplified by drought, inhibiting the photosynthetic activity.Citation16 This effect is encountered by a desiccation-related ELIP gene, which is regulated by light and ABA.Citation32 The expression of the ELIP gene family has higher expression during drought stress in resurrection plant S. lepidophyllaCitation33 and B. hygrometrica.Citation34 According to,Citation35 ELIPs showed low expression in rehydrated tissues and helped plants resynthesize chlorophyll. Expression of ELIP transcripts was found in one of the moss species Syntrichis during environmental stress.Citation36,Citation37 When plants return to normal water content, desiccation-tolerant species show decreased expression of ELIPs.Citation35 We obtained similar results in our study, the unigenes encoding ELIPs showed lower expression during the fully watered condition of S. tamariscina leaves (), indicating that lycophyll in dehydration is more likely to protect chlorophyll.
Pentatricopeptide repeat-containing proteins (PPRs) are involved in ABA signaling and play an important role in drought tolerance, cold stress and salinity.Citation38 In response to rehydration, there is high expression of PPR genes in S. tamariscina, in which plants are involved in chloroplast development.Citation17 In Arabidopsis, up regulation of PPR gene negatively regulates NADH dehydrogenase activity and enhance defense mechanism under abiotic stress.Citation38,Citation39 found up regulation of PPR protein SOAR1 enhances ABA sensitivity, and overexpression of this gene strongly increases the drought tolerance ability of Arabidopsis. In this study, we identified a high number of up regulated DEGs under rehydration that belong to the PPR gene family (). This indicates that PPR genes might play a potential role in rehydrating plants from dehydration through maintenance and development of chloroplast.
In plants, LEA proteins are well-known ion scavengers,Citation40 which function in reducing oxidative damage generated by abiotic stress in soybeans.Citation29 Significant up regulation of LEA genes protects the plants during drought stress.Citation17 Overexpression of transgenic rice and wheat LEA genes are regulated by ABA which resulted drought tolerance.Citation41 Similarly, overexpression of the Oryza sativa LEA gene improved drought resistance with high yield in field conditions.Citation42 In our study, the LEA gene family are up regulated in rehydration, which shows its important role in the protection and regeneration of plants after dehydration. Sucrose non-fermenting (SNF) proteins have been widely studied in several plants and play an important role in physiological resistance.Citation43 In Arabidopsis, SNF4 regulates ROS in pollen and helps pollen hydration.Citation44 Overexpression of SNF-related kinase 2 in transgenic tobacco improved drought stress and increased the survival rate through an improved antioxidant system.Citation45 The expression of SNF genes is increased during hydration, which might help in the regeneration of carbohydrate metabolism and starch biosynthesis in plants.Citation46 We found similar result in our study, unigenes encoding SNF proteins are up regulated during rehydration.
Trehalose is a disaccharide sugar consisting of two glucose molecules that functions in sugar transport during dehydration.Citation47 Trehalose is known to exert a strengthening effect on biological structures by forming a glass-like structure after dehydration.Citation48 The trehalose synthase complex is involved in the formation of trehalose from the substrate UDP-glucose.Citation49,Citation50 TPS encodes the enzyme trehalose-6-phosphate synthase, which catalyzes the conversion of UDP-glucose to trehalose-6-phosphate, and TPP encodes the enzyme trehalose-6-phosphate phosphatase, which catalyzes the conversion of trehalose-6-phosphate to trehalose (). During dehydration, TPS1 and TPP4 play significant roles in stabilizing proteins in plants, which helps during dehydration. The mutant of Arabidopsis lacking the TPP gene resulted in a drought-sensitive phenotype, and overexpression of the same gene increased the drought tolerance.Citation51 Zentella et al.Citation52 demonstrated that TPS1 mRNA was constitutively expressed in Selaginella lepidophylla, which is known as a resurrection plant. When S. lepidophylla TPS1 and SlTPS1 were introduced to yeast, the transformed yeast showed tolerance at high temperatures. In Arabidopsis, Citation50,reported that overexpression of AtTPS1 displayed dehydration tolerance [Citation53]; on this basis, they posited that trehalose-6-P synthase involving AtTPS1 plays a pivotal role in the regulation of glucose and ABA signaling during vegetative development.Citation54 In our results, we found that TPP and TPS coding unigenes are upregulated in the fully rehydration condition.
ABC transporter are one of the largest and oldest protein families in prokaryotes and eukaryotes.Citation24 The G sub-family of ABC transporters is the largest known family in the context of protein structure.Citation55 In pathway analysis, ABCG genes are found in purine metabolism and thiamine metabolism, which converts thiamine diphosphate to thiamine phosphate ( b, c). Thiamine metabolism was modulated under the condition of abiotic stress in Zea mays seedlings.Citation56 ABCG genes are essential for vascular development in A. thaliana.Citation57 Overexpression of ABCG25 gene in A. thaliana reduced the rate of water loss, indicating that AtABCG25 facilitates ABA in guard cell-enhancing stomata closure.Citation14 The mutant of abcg40 in Arabidopsis reduced the role of ABA, and plants were found to be more susceptible to drought stress.Citation58 In the present study, the ABCG gene showed up regulation during rehydration, indicating that ABCG might play an important role in the deregulation of stomata opening during resurrection.
Conclusion
Using the Illumina platform, we analyzed the gene expression of dehydrated and rehydrated lycohyll of S. tamaricina. Comparative gene expression identified 1901 DEGs involved in resurrection. More number of DEGs were upregulated in rehydration compared to dehydration. The selected genes are mostly involved in ABA hormone signaling and play important roles in drought tolerance – especially in chloroplast protection, reducing oxidative damage, accumulation of sucrose and trehalose, and vascular development in plants under the acquired environmental period (dehydration and rehydration). The up regulation of these genes relates to increased tolerance to desiccation. In this study, we provide the most promising resurrection genes and their functions that could be improved biotechnologically to obtain drought-tolerant plants.
Authors’ contributions
N.-SK and I.-YC designed the project and wrote the manuscript. EK, J.-HK and KH prepared the samples and analyzed the data. K.-CP, PB and TU analyzed the data and performed the wet experiment.
Supplemental Material
Download Zip (200.5 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
Additional information
Funding
Related Research Data
References
- Thomson WW. Conservation of Cell Order in Desiccated Mesophyll ofSelaginella lepidophylla([Hook and Grev.] Spring). Annals of Botany. 1997;79(4):1–9. doi:https://doi.org/10.1371/journal.pgen.1006228.
- Rajeev L, Da Rocha UN, Klitgord N, Luning EG, Fortney J, Axen SD, Shih PM, Bouskill NJ, Bowen BP, Kerfeld CA, et al. Dynamic cyanobacterial response to hydration and dehydration in a desert biological soil crust. ISME J. 2013;7(11):2178–2191. doi:https://doi.org/10.1038/ismej.2013.83.
- Zaremba LS, Smoleński WH. Optimal portfolio choice under a liability constraint. Annals of Operations Research. 2000;97(1/4):131–141. doi:https://doi.org/10.1023/A:1018996712442.
- Porembski S, Barthlott W. Granitic and gneissic outcrops (inselbergs) as centers of diversity for desiccation-tolerant vascular plants. Plant Ecol. 2000;151(1):19–28. doi:https://doi.org/10.1023/A:1026565817218.
- Rascio N, La Rocca N. Resurrection plants: the puzzle of surviving extreme vegetative desiccation. CRC Crit Rev Plant Sci. 2005;24(3):209–225. doi:https://doi.org/10.1080/07352680591008583.
- Yobi A, Wone BW, Xu W, Alexander DC, Guo L, Ryals JA, Oliver MJ, Cushman JC. Metabolomic profiling in selaginella lepidophylla at various hydration states provides new insights into the mechanistic basis of desiccation tolerance. Mol. Plant. 2013;6(2):369–385. doi:https://doi.org/10.1093/mp/sss155.
- Oliver MJ. Desiccation tolerance in vegetative plant cells. Physiologia Plantarum. 1996;97(4):779–787. doi:https://doi.org/10.1104/pp.104.052084.
- Bartels D, Hussain SS. Resurrection plants: physiology and molecular biology. 2011. 339–364. doi:https://doi.org/10.1007/978-3-642-19106-0_16.
- Lebkuecher JG, Eickmeier WG. Reduced photoinhibition with stem curling in the resurrection plant Selaginella lepidophylla. Oecologia. 1991;88(4):597–604. doi:https://doi.org/10.1007/BF00317725.
- Farrant JM. Uma comparação dos padrões de tolerância à dessecação entre três espécies de plantas angiospermas ressurreição. Plant Ecol. 2000 in press;151(1):29–39. doi:https://doi.org/10.1023/A:1026534305831.
- Oliver MJ, Velten J, Mishler BD. Desiccation tolerance in bryophytes: a reflection of the primitive strategy for plant survival in dehydrating habitats? Integr. Comp. Biol. 2005;45(5):788–799. doi:https://doi.org/10.1093/icb/45.5.788.
- Tuba Z, Protor MCF, Csintalan Z. Ecophysiological responses of homoiochlorophyllous and poikilochlorophyllous desiccation tolerant plants: a comparison and an ecological perspective. Plant Growth Regul. 1998;24(3):211–217. doi:https://doi.org/10.1023/A:1005951908229.
- Liu Y, He J, Chen Z, Ren X, Hong X, Gong Z. ABA overly-sensitive 5 (ABO5), encoding a pentatricopeptide repeat protein required for cis-splicing of mitochondrial nad2 intron 3, is involved in the abscisic acid response in Arabidopsis. Plant J. 2010;63(5):749–765. doi:https://doi.org/10.1111/j.1365-313X.2010.04280.x.
- Kuromori T, Miyaji T, Yabuuchi H, Shimizu H, Sugimoto E, Kamiya A, Moriyama Y, Shinozaki K. ABC transporter AtABCG25 is involved in abscisic acid transport and responses. Proc Natl Acad Sci U S A. 2010;107(5):2361–2366. doi:https://doi.org/10.1073/pnas.0912516107.
- Casazza AP, Rossini S, Rosso MG, Soave C. Mutational and expression analysis of ELIP1 and ELIP2 in Arabidopsis thaliana. Plant Mol. Biol. 2005;58(1):41–51. doi:https://doi.org/10.1007/s11103-005-4090-1.
- Hutin C, Nussaume L, Moise N, Moya I, Kloppstech K, Havaux M. Early light-induced proteins protect Arabidopsis from photooxidative stress. Proc Natl Acad Sci U S A. 2003;100(8):4921–4926. doi:https://doi.org/10.1073/pnas.0736939100.
- Xu Z, Xin T, Bartels D, Li Y, Gu W, Yao H, Liu S, Yu H, Pu X, Zhou J, et al. Genome analysis of the ancient tracheophyte selaginella tamariscina reveals evolutionary features relevant to the acquisition of desiccation tolerance. Mol. Plant. 2018;11(7):983–994. doi:https://doi.org/10.1016/j.molp.2018.05.003.
- Ramos-Vega M, Guevara-García A, Llamas E, Sánchez-León N, Olmedo-Monfil V, Vielle-Calzada JP, León P. Functional analysis of the Arabidopsis thaliana CHLOROPLAST BIOGENESIS 19 pentatricopeptide repeat editing protein. New Phytol. 2015;208(2):430–441. doi:https://doi.org/10.1111/nph.13468.
- Dahal K, Vanlerberghe GC. Alternative oxidase respiration maintains both mitochondrial and chloroplast function during drought. New Phytol. 2017;213(2):560–571. doi:https://doi.org/10.1111/nph.14169.
- Tolleter D, Jaquinod M, Mangavel C, Passirani C, Saulnier P, Manon S, Teyssier E, Payet N, Avelange-Macherel MH, Macherel D. Structure and function of a mitochondrial late embryogenesis abundant protein are revealed by desiccation. Plant Cell. 2007;19(5):1580–1589. doi:https://doi.org/10.1105/tpc.107.050104.
- Avonce N, Mendoza-Vargas A, Morett E, Iturriag G. Insights on the evolution of trehalose biosynthesis. BMC Evol. Biol. 2006;6(1):1–15. doi:https://doi.org/10.1186/1471-2148-6-109.
- Ponnu J, Wahl V, Schmid M. Trehalose-6-phosphate: connecting plant metabolism and development. Frontiers in Plant Science. 2011;2:1–6. doi:https://doi.org/10.3389/fpls.2011.00070.
- Carianopol CS, Chan AL, Dong S, Provart NJ, Lumba S, Gazzarrini S. An abscisic acid-responsive protein interaction network for sucrose non-fermenting related kinase1 in abiotic stress response. Commun. Biol. 2020;3(1):1–15. doi:https://doi.org/10.1038/s42003-020-0866-8.
- Hwang JU, Song WY, Hong D, Ko D, Yamaoka Y, Jang S, Yim S, Lee E, Khare D, Kim K, et al. Plant ABC transporters enable many unique aspects of a terrestrial plant’s lifestyle. Mol. Plant. 2016;9(3):338–355. doi:https://doi.org/10.1016/j.molp.2016.02.003.
- Moore JP, Le NT, Brandt WF, Driouich A, Farrant JM. Towards a systems-based understanding of plant desiccation tolerance. Trends Plant Sci. 2009 Feb;14(2):110–117. doi:https://doi.org/10.1016/j.tplants.2008.11.007.
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011 May 15;29(7):644–652. doi: https://doi.org/10.1038/nbt.1883.
- Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012 Dec 1;28(23):3150–3152. https://doi.org/10.1093/bioinformatics/bts565.
- Mulder N, Apweiler R. InterPro and InterProScan: tools for protein sequence classification and comparison. Methods Mol. Biol. 2007;396:59–70. https://doi.org/10.1007/978-1-59745-515-2-5.
- Liu G, Xu H, Zhang L, Zheng Y. Fe binding properties of two soybean (Glycine max L.) LEA4 proteins associated with antioxidant activity. Plant Cell Physiol. 2011;52(6):994–1002. doi:https://doi.org/10.1093/pcp/pcr052.
- Sun J, Nishiyama T, Shimizu K, Kadota K. TCC: an R package for comparing tag count data with robust normalization strategies. BMC Bioinformatics. 2013 Jul 9;14:219. https://doi.org/10.1186/1471-2105-14-219.
- Young MD, Wakefield MJ, Smyth GK, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11(2):R14. https://doi.org/10.1186/gb-2010-11-2-r14.
- Bartels D, Hanke C, Schneider K, Michel D, Salamini F. A desiccation-related Elip-like gene from the resurrection plant Craterostigma plantagineum is regulated by light and ABA. EMBO J. 1992;11(8):2771–2778. doi:https://doi.org/10.1002/j.1460-2075.1992.tb05344.x.
- VanBuren R, Pardo J, Man Wai C, Evans S, Bartels D. Massive tandem proliferation of ELIPs supports convergent evolution of desiccation tolerance across land plants. Plant Physiol. 2019 Mar;179(3):1040–1049. https://doi.org/10.1104/pp.18.01420.
- Xiao L, Yang G, Zhang L, Yang X, Zhao S, Ji Z, Zhou Q, Hu M, Wang Y, Chen M, et al. The resurrection genome of Boea hygrometrica : a blueprint for survival of dehydration. Proc Natl Acad Sci U S A. 2015;112(18):5833–5837. doi:https://doi.org/10.1073/pnas.1505811112.
- Van Buren R, Pardo J, Wai CM, Evans S, Bartels D. Massive tandem proliferation of elips supports convergent evolution of desiccation tolerance across land plants. Plant Physiol. 2019;179(3):1040–1049. doi:https://doi.org/10.1104/pp.18.01420.
- Oliver MJ, Dowd SE, Zaragoza J, Mauget SA, Payton PR. The rehydration transcriptome of the desiccation-tolerant bryophyte Tortula ruralis: transcript classification and analysis. BMC Genomics. 2004;5(1):1–19. doi:https://doi.org/10.1186/1471-2164-5-89.
- Zeng Q, Chen X, Wood AJ. Two early light-inducible protein (ELIP) cDNAs from the resurrection plant Tortula ruralis are differentially expressed in response to desiccation, rehydration, salinity, and high light. J. Exp. Bot. 2002;53(371):1197–1205. doi:https://doi.org/10.1093/jexbot/53.371.1197.
- Jiang SC, Mei C, Liang S, Yu YT, Lu K, Wu Z, Wang XF, Zhang DP. Crucial roles of the pentatricopeptide repeat protein SOAR1 in Arabidopsis response to drought, salt and cold stresses. Plant Mol. Biol. 2015;88(4–5):369–385. doi:https://doi.org/10.1007/s11103-015-0327-9.
- Yuan H, Liu D. Functional disruption of the pentatricopeptide protein SLG1 affects mitochondrial RNA editing, plant development, and responses to abiotic stresses in Arabidopsis. Plant J. 2012;70(3):432–444. doi:https://doi.org/10.1007/BF00317725.
- Imai R, Chang L, Ohta A, Bray EA, Takagi M. A lea-class gene of tomato confers salt and freezing tolerance when expressed in Saccharomyces cerevisiae. Gene. 1996 May 8;170(2):243–248. https://doi.org/10.1016/0378-1119(95)00868-3.
- Chen Y, Li C, Zhang B, Yi J, Yang Y, Kong C, Lei C, Gong M. The role of the Late Embryogenesis-Abundant (LEA) protein family in development and the abiotic stress response: a comprehensive expression analysis of potato (Solanum Tuberosum). Genes (Basel). 2019;10(2):1–16. doi:https://doi.org/10.3390/genes10020148.
- Xiao B, Huang Y, Tang N, Xiong L. Over-expression of a LEA gene in rice improves drought resistance under the field conditions. Theor Appl Genet. 2007;115(1):35–46. doi:https://doi.org/10.1007/s00122-007-0538-9.
- He H, Qin Z, Feng Z, Wu T. Cloning and expression analysis of sucrose non-fermenting-1 related protein kinase (SnRK) in cucumis sativus L. under low nitrogen conditions. J. Northeast Agric. Univ. (English Ed. 2013;20(4):1–9. doi:https://doi.org/10.1016/s1006-8104(14)60040-4.
- Gao XQ, Liu CZ, Li DD, Zhao TT, Li F, Jia XN, Zhao XY, Zhang XS. The arabidopsis KINβγ subunit of the SnRK1 complex regulates pollen hydration on the stigma by mediating the level of reactive oxygen species in pollen. PLoS Genet. 2016;12(7):1–25. doi:https://doi.org/10.1371/journal.pgen.1006228.
- Feng J, Wang L, Wu Y, Luo Q, Zhang Y, Qiu D, Han J, Su P, Xiong Z, Chang J, et al. TaSnRK2.9, a sucrose non-fermenting 1-related protein kinase gene, positively regulates plant response to drought and salt stress in transgenic tobacco. Frontiers in Plant Science. 2019;9:1–17. doi:https://doi.org/10.3389/fpls.2018.02003.
- Wang F, Ren G, Li F, Wang B, Yang Y, Ma X, Niu Y, Ye Y, Chen X, Fan S, et al. Overexpression of GmSnRK1, a soybean sucrose non-fermenting-1 related protein kinase 1 gene, results in directional alteration of carbohydrate metabolism in transgenic Arabidopsis. Biotechnol. Biotec. Equip. 2018;32(4):835–845. doi:https://doi.org/10.1146/annurev.cb.08.110192.000435.
- Wiemken A. Trehalose in yeast, stress protectant rather than reserve carbohydrate. Antonie Van Leeuwenhoek. 1990;58(3):209–217. doi:https://doi.org/10.1007/BF00548935.
- Kaushik JK, Bhat R. Why is trehalose an exceptional protein stabilizer? An analysis of the thermal stability of proteins in the presence of the compatible osmolyte trehalose. J. Biol. Chem. 2003 Jul 18;278(29):26458–26465. https://doi.org/10.1074/jbc.M300815200.
- Goddijn OJ, van Dun K. Trehalose metabolism in plants. Trends Plant Sci. 1999 Aug;4(8):315–319. doi:https://doi.org/10.1016/s1360-1385(99)01446-6.
- Avonce N, Leyman B, Mascorro-Gallardo JO, van Dijck P, Thevelein JM, Iturriaga G. The arabidopsis trehalose-6-P synthase AtTPS1 gene is a regulator of glucose, abscisic acid, and stress signaling. Plant Physiol. 2004;136(3):3649–3659. doi:https://doi.org/10.1104/pp.104.052084.
- Lin Q, Wang S, Dao Y, Wang J, Wang K. Arabidopsis thaliana trehalose-6-phosphate phosphatase gene TPPI enhances drought tolerance by regulating stomatal apertures. J. Exp. Bot. 2020;71(14):4285–4297. doi:https://doi.org/10.1093/jxb/eraa173.
- Zentella R, Mascorro-Gallardo JO, Van Dijck P, Folch-Mallol J, Bonini B, Van Vaeck C, Gaxiola R, Covarrubias AA, Nieto-Sotelo J, Thevelein JM, et al. A Selaginella lepidophylla trehalose-6-phosphate synthase complements growth and stress-tolerance defects in a yeast tps1 mutant. Plant Physiol. 1999 Apr;119(4):1473–1482. doi:https://doi.org/10.1104/pp.119.4.1473.
- Sassi M, Ali O, Boudon F, Cloarec G, Abad U, Cellier C, Chen X, Gilles B, Milani P, Friml J, et al. An auxin-mediated shift toward growth isotropy promotes organ formation at the shoot meristem in arabidopsis. Curr Biol. 2014;24(19):2335–2342. doi:https://doi.org/10.1016/j.cub.2014.08.036.
- Higgins CF. ABC Transporters: from microorganisms to man. Annu. Rev. Cell Biol. 1992;8(1):67–113. doi:https://doi.org/10.1146/annurev.cb.08.110192.000435.
- Jarzyniak KM, Jasiå„ski M. Membrane transporters and drought resistance – a complex issue. Frontiers in Plant Science. 2014;5:1–16. doi:https://doi.org/10.3389/fpls.2014.00687.
- Rapala-Kozik M, Kowalska E, Ostrowska K. Modulation of thiamine metabolism in Zea mays seedlings under conditions of abiotic stress. J. Exp. Bot. 2008;59(15):4133–4143. doi:https://doi.org/10.1093/jxb/ern253.
- Le Hir R, Sorin C, Chakraborti D, Moritz T, Schaller H, Tellier F, Robert S, Morin H, Bako L, Bellini C. ABCG9, ABCG11 and ABCG14 ABC transporters are required for vascular development in Arabidopsis. Plant J. 2013;76(5):811–824. doi:https://doi.org/10.1111/tpj.12334.
- Kang J, Hwang JU, Lee M, Kim YY, Assmann SM, Martinoia E, Lee Y. PDR-type ABC transporter mediates cellular uptake of the phytohormone abscisic acid. Proc Natl Acad Sci U S A. 2010;107(5):2355–2360. doi:https://doi.org/10.1073/pnas.0909222107.