
ABSTRACT
Platycodon grandiflorus, a perennial flowering plant widely distributed in China and South Korea, is an excellent resource for both food and medicine. The main active compounds of P. grandiflorus are triterpenoid saponins. WRKY transcription factors (TFs) are among the largest gene families in plants and play an important role in regulating plant terpenoid accumulation, physiological metabolism, and stress response. Numerous studies have been reported on other medicinal plants; however, little is known about WRKY genes in P. grandiflorus. In this study, 27 PgWRKYs were identified in the P. grandiflorus transcriptome. Phylogenetic analysis showed that PgWRKY genes were clustered into three main groups and five subgroups. Transcriptome analysis showed that the PgWRKY gene expression patterns in different tissues differed between those in Tongcheng City (Southern Anhui) and Taihe County (Northern Anhui). Gene expression analysis based on RNA sequencing and qRT-PCR analysis showed that most PgWRKY genes were expressed after induction with methyl jasmonate (MeJA). Co-expressing PgWRKY genes with triterpenoid biosynthesis pathway genes revealed four PgWRKY genes that may have functions in triterpenoid biosynthesis. Additionally, functional annotation and protein-protein interaction analysis of PgWRKY proteins were performed to predict their roles in potential regulatory networks. Thus, we systematically analyzed the structure, evolution, and expression patterns of PgWRKY genes to provide an important theoretical basis for further exploring the molecular basis and regulatory mechanism of WRKY TFs in triterpenoid biosynthesis.
1 Introduction
The dried roots of Platycodon grandiflorus (Jacq.) A.DC is commonly used as a traditional medicine across northern and southern regions of China.Citation1 P. grandiflorus contains triterpenoid saponins, polysaccharides, flavonoids, phenols, sterols, polyalkynes, fatty acids, trace elements, and other chemical components, among which triterpenoid saponins are the main active compounds.Citation2,Citation3 P. grandiflorus also has various pharmacological effects, including anti-inflammatory,Citation4,Citation5 anti-tumor,Citation6,Citation7 and hepatoprotective Citation8,Citation9 effects. Owing to changes in the environment and climate, the chemical content of P. grandiflorus varies in different regions. At the same time, the growth and development of plants are also regulated by a variety of hormones. Among them, methyl jasmonate (MeJA) is widely present in plants and activates the coordinated expression of a series of response genes through the transduction of JA signaling, thereby regulating plant growth and development, stress response and secondary metabolism.Citation10–13 For example, MeJA can increase the content of artemisinin by increasing the expression level of AaWRKY9.Citation14 LrWRKY4 and LrWRKY12 were responded to MeJA and enhanced Lilium regale resistance to gray mold.Citation15 PpWRKY46 and PpWRKY53 can interact to regulate MeJA-mediated energy metabolism in peach disease resistance.Citation16
WRKY TFs are one of the largest TF families in plants. They contain a highly conserved WRKY motif that regulates various physiological processes, including stress, growth, and development, thus forming a complete and complex molecular signaling network.Citation17,Citation18 These factors feature a DNA-binding domain, a highly conserved WRKY motif consisting of 60 amino acids (aa) with a highly-conserved N-terminal WRKYGQK polypeptide sequence, and a C-terminal zinc finger motif, which can be classified into C2H2 and C2HC types.Citation19,Citation20 Based on the number of WRKY TF structural domains and the zinc finger structure type, WRKY TFs are classified into three categories:Citation21 Class I contains two WRKY motifs with a C2H2-type zinc finger; Class II contains one WRKY motif with a C2H2-type zinc finger structure, which can be further divided into five subgroups named IIa, -b, -c, -d, and -e based on phylogenetic relationships; and Class III contains one WRKY motif with a C2HC-type zinc finger.Citation22–24
The WRKY transcription factor family was first identified as SPF1 in the cDNA of sweet potatoes (Lpomoea batatas) in 1994.Citation25 Over time, WRKY TFs have been discovered various plants, such as Arabidopsis thaliana,Citation26 Oryza sativa,Citation27 Phakopsora pachyrhizi,Citation28 and Panax ginseng.Citation29 WRKY TFs regulate plant growth and development, secondary metabolic pathways, and responses to biotic and abiotic stressors.Citation30 Recent studies have suggested that WRKY TFs in help the seed germination,Citation31 the formation and development of plant roots, stems, and leaves,Citation32 as well as growth and development, including reproduction Citation33,Citation34 and senescence.Citation35,Citation36 WRKY TFs also function in secondary metabolic pathways of several medicinal plants. For example, the AaWRKY1 gene in Artemisia annua has been shown to regulate of artemisinin biosynthesis,Citation37 and overexpression of the PqWRKY1 gene activates the triterpene biosynthetic pathway in Panax quinquefolius.Citation34 WRKY are also indispensable to abiotic and biotic stresses, which enhance the resistance of Arabidopsis to pathogens Citation38,Citation39 and increase the tolerance of plants to drought,Citation40,Citation41 high temperatures,Citation42,Citation43 low temperatures,Citation44,Citation45 and other stresses.
Our research has shown that the content of P. grandiflorus triterpenoids from the southern regions of China is higher than that of the northern regions.Citation46–48 We also identified the transcriptomes of P. grandiflorus in combination with PacBio and DNBSEQ sequencing platforms.Citation49 The WRKY gene family, a critical transcriptional factor in regulating secondary metabolism and stress response in plants, plays a vital role in the growth and development of P. grandiflorus. However, only a few studies have been performed on the WRKY gene family of P. grandiflorus. The expression trends of this gene family in different parts of P. grandiflorus and their response to abiotic stress remain unclear.
We conducted a comprehensive investigation of the WRKY gene family in P. grandiflorus. We identified 27 P. grandiflorus WRKY genes and analyzed sequence alignments, conserved motifs, phylogenetic trees, GO annotations, and protein interactions. RNA-sequencing (RNA-seq) data were used to study the expression profile of PgWRKY genes in different tissues and their response trends under MeJA treatment. The expression of selected PgWRKY genes under MeJA treatment was verified by quantitative real-time PCR (qRT-PCR). Based on the tissue-specific expression of the PgWRKY gene and co-expression of genes involved in the triterpenoid biosynthesis pathway of P. grandiflorus, we proposed the potential regulatory TFs involved in the triterpenoid biosynthesis in P. grandiflorus, which laid a foundation for further study of the WRKY gene family in P. grandiflorus.
2 Materials and methods
2.1 Plant materials and sample collection
Biennial Platycodon grandiflorus were collected from Tongcheng City (Southern Anhui) and Taihe County (Northern Anhui) respectively, and were identified as P. grandiflorus by Professor Peng Huasheng. It was separated into roots, stems, and leaves with three biological replicates per organ. After a quick rinse with sterile water, samples were snap frozen in liquid nitrogen and stored at −80°C for subsequent analysis.
The material used for hormone treatment was Tongcheng P. grandiflorus tissue culture seedlings provided by College of Pharmacy, Anhui University of Chinese Medicine, Hefei, China, and treated with 50 μM MeJA for 0, 3, 6, 9, 12, 24 and 48 h. Root tissues of plants were harvested, all treatments were three biological replicates. Freeze immediately in liquid nitrogen and store at −80°C.
2.2 Analysis of saponin contents in P. grandiflorus
The root, stem and leaf tissues were dried under 55°C, ground into powder and passed through a 50-mesh sieve. 10 mg powder samples were weighed and extracted with 8 mL of 70% methanol by ultrasonic extraction for 1 h (100 W, 40 Hz). The extracted samples were passed through 0.45 μm filter membrane and injected into the column.Citation46 The contents of eight Platycodon saponins were determined by ultra-high performance liquid chromatography-Orbitrap-tandem mass spectrometry (UHPLC-Orbitrap-MS/MS). All analyses were performed on an Ultimate HPLC system (Thermo Fisher Scientific, Waltham, MA, United States). The samples were separated on an Agilent Eclipse XDB-C18 column (4.6 mm × 250 mm, 5 μm). The mobile phase was 0.075% acetic acid water (A)-methanol (B). The gradient curves were as follows: 0 ~ 3 min, 90 ~ 80% A; 3 ~ 11 min, 80 ~ 77% A; 11 ~ 20 min, 77 ~ 5% A; 20 ~ 25 min, 5 ~ 90% A. The mobile phase flow rate was 0.3 mL/min, and the injection volume was 2 μL.
2.3 Identification of WRKY Gene family in P. grandiflorus
Based on the existing transcriptome sequencing data of the research group (SRA accession number: PRJNA688328), all presumed WRKY proteins of P. grandiflorus were retrieved by searching against the transcriptome annotation sequence. The sequences with complete WRKY domains were selected, and the homology of amino acid sequence above 97 % were deleted. The amino acid sequences of WRKY in A. thaliana published on the Arabidopsis Information Resource website (http://www.arabidopsis.org/) were downloaded and aligned to remove sequences with incomplete WD via the MEGA-X. Subsequently, all validated candidate genes with probable WRKY domains were corroborated using the SMART program (http://smart.embl-heidelberg.de) to obtain sequences with the WRKY domain only. The putative WRKY genes from P. grandiflorus were named PgWRKY1-PgWRKY27 based on their relationship with the A. thaliana sequence. The isoelectric points (pIs) and theoretical molecular weights (MWs) of each PgWRKY protein were predicted online using the Expasy portal (https://web.expasy.org/protparam/). Cell localization was predicted using the WoLF PSORT tool (https://wolfpsort.hgc.jp/).
2.4 Multiple sequence alignment and protein structure analysis
Accurately grouping gene families is an integral part of the functional analysis of transcription factors. Based on the AtWRKYs classification in A. thaliana, 72 AtWRKYs were downloaded from the Arabidopsis Information Resource (https://www.arabidopsis.org/), and all identified PgWRKYs were divided into different groups according to the classification of AtWRKYs. Multiple sequence alignment of domains in PgWRKYs was executed using the ClustalW program with default parameters,Citation50 and the results were colored using the GeneDoc tool. The conserved motifs of PgWRKYs were analyzed using multiple expectation maximization for motif elicitation (MEME: http://meme-suite.org/tools/meme) with the following parameters: repetitive time was “any,” maximum motif number was 10, and motif width was between 6 and 50 residues.Citation51,Citation52 The MEME results were displayed using TBtool software.Citation53
2.5 Phylogenetic analysis of PgWRKY
The amino acid sequences of WRKY in P. grandiflorus and A. thaliana were aligned using ClustalW,Citation50 and then imported to construct an evolutionary relationship tree using the maximum likelihood method.Citation54 The substitution model, equal input model, rates among sites, uniform rates, gaps, missing data treatment, and partial deletion parameters were used in phylogenetic tree construction. The trees were visualized and optimized using the iTOL tool (https://itol.embl.de/).
2.6 Expression profile analysis of PgWRKY genes
We analyzed the expression of WRKY genes in different tissues of P. grandiflorus and their response to MeJA treatment. Our group completed transcriptome sequencing of different tissues of P. grandiflorus (SRA accession number: PRJNA688328), and 12 RNA-Seq datasets for MeJA processing were obtained from NCBI (SRX5506849, SRX5506850, SRX5506841, SRX5506842, SRX5506839, SRX5506840, SRX5506845, SRX5506846, SRX5506843, SRX56847, SRX550656847, SRX55065684RX). Clean reads obtained from RNA-seq were mapped to the reference full-length transcripts using the Bowtie2 tool.Citation55 The expression levels were then calculated for each sample using RNA-seq by expectation-maximization (RSEM) software, and the transcript per kilobase fragment mapping (FPKM) was used to normalize the read count.Citation56 To analyze the expression of genes related to triterpenoid synthesis pathway in P. grandiflorus, we obtained KEGG annotations from transcriptome data (SRA accession number: PRJNA688328). We screened genes related to synthesis of platycodins according to annotation information of map00900 (terpenoid backbone biosynthesis) and map00909 (sesquiterpenoid and triterpenoid biosynthesis) in KEGG database.Citation49 The differentially expressed genes (DEGs) in different tissues of roots, stems and leaves were further screened according to the their FPKM values. A total of 7 DEGs encoding 6 enzymes related to the MVA pathway, 8 DEGs encoding 6 enzymes involved in the MEP pathway, and 9 DEGs of 5 enzymes of the downstream of triterpenoid saponin synthesis pathway were identified. Pearson correlations between genes from the P. grandiflorus triterpenoid biosynthetic pathway and PgWRKYs of Tongcheng P. grandiflorus were calculated using the SPSS software. Heat maps were plotted using the TBtools software.Citation53
2.7 Gene expression analysis by quantitative real-time PCR (qRT-PCR)
Total RNA was extracted using RNAiso Plus (TaKaRa, Japan), according to the manufacturer’s instructions. RNA (1 μg) was reverse-transcribed to cDNA using a reverse transcription kit (PrimeScript™ II 1st Strand cDNA Synthesis Kit, Waryong, China). qRT-PCR was performed to determine the relative expression levels of PgWRKY genes under MeJA treatment, and β-actin was used as an internal control. Primer sequences are listed in Supplementary Table 1. The reaction system was prepared according to the instructions of the TB Green® Premix Ex Taq™ II kit. The PCR conditions were as follows: 95°C for 30s, 95°C for 15s, 60°C for 30s, 72°C for 15s, and 40 cycles. Three biological replicates and three technical replicates were set up, melting curves were plotted, and the relative gene expression levels were analyzed using the 2−ΔΔCt method.Citation57
2.8 Gene ontology annotation and protein-protein interaction analysis
Gene ontology (GO) annotation was obtained from Beijing Genomics Institute (BGI; https://report.bgi.com/ps/login/login.html) and visualized using OmicShare (https://www.omicshare.com/tools/). The STRING 11.5 (http://string-db.org/) database was used to predict Arabidopsis WRKY protein-protein interaction networks.Citation58 Fourteen AtWRKY proteins were identified, with a confidence parameter of 0.4. AtWRKY proteins with interactions were mapped to PgWRKY proteins by homology.
3 Results
3.1 Identification of WRKY Gene family in P. grandiflorus
Twenty-seven candidate PgWRKY genes were identified in the full-length transcriptome of P. grandiflorus Citation49 using AtWRKY genes in A. thaliana as queries. We examined the sequences of all candidate proteins using the conserved domain database (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi). We also verified that all 27 PgWRKY proteins contained the WRKY DNA-binding domain. As shown in , all the identified PgWRKY proteins contained at least one highly conserved heptapeptide WRKY domain; however, a difference was found in the WRKYGQK motif and the zinc-finger-like motif. Further analysis indicated that 26 PgWRKY proteins contain the conserved WRKYGQK heptapeptide domain, and the PgWRKY10 protein contains a WRKYGKK sequence. The ORF length for the PgWRKY genes ranged from 576 bp (PgWRKY10) to 2196 bp (PgWRKY2), encoding 191–731 aa. The deduced 27 PgWRKY proteins have MWs from 21.29 kDa (PgWRKY10) to 78.99 kDa (PgWRKY2) and pIs from 4.91 (PgWRKY16) to 9.66 (PgWRKY24). The subcellular localization prediction showed that most members (except PgWRKY17) were present in the nucleus. Supplementary Table 2 contains specific information on the WRKY genes of P. grandiflorus.
Table 1. Features of PgWRKY genes identified in P. grandiflorus.
3.2 Multiple sequence alignment and structure analysis
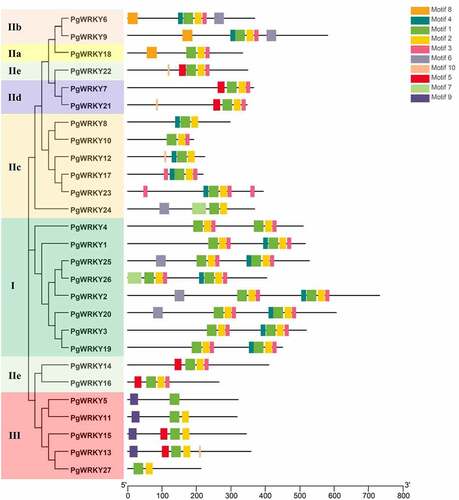
Seven Arabidopsis proteins from different groups were randomly selected as representative sequences (Group I: AtWRKY58, Group IIa: AtWRKY40, Group IIb: AtWRKY6, Group IIc: AtWRKY56, Group IId: AtWRKY21, Group IIe: AtWRKY35, and Group III: AtWRKY46). The detailed structures of the WRKY domain and zinc-finger type are displayed in . The WRKYGQK heptapeptide is a signature of WRKY proteins. Twenty-six PgWRKY proteins contained the highly conserved WRKYGQK sequence, while PgWRKY10 had a single amino acid substitution, K, for Q ( and ). The second motif is a zinc-finger structure containing two types of zinc finger motifs: C-X4-5-C-X22-23-H-X-H (C2H2) and C-X7-C-X23-H-X-C (C2HC). Twenty-one PgWRKY proteins included C2H2 type zinc finger motifs, five PgWRKY proteins displayed C2HC type zinc finger motifs, and a partial absence of the zinc-finger motif sequence was present in PgWRKY24. A comparison of the WRKY TFs identified in the P. grandiflorus transcriptome is presented in Supplementary Figure 1.
Figure 1. Alignment of 27 P. grandiflorus (PgWRKY) and 8 A. thaliana (AtWRKY) WRKY domain sequences. For group I WRKY proteins, N-terminal and C-terminal WRKY domains are represented by N and C, respectively. In the red solid box are WRKY domains. Zinc finger structures are marked by a red dotted box.

To gain insight into the functional regions of PgWRKY proteins, the MEME program was employed to reveal conserved motifs among 27 PgWRKY proteins. The MEME results showed that the number of motifs in the PgWRKYs ranged from 2–8 aa, and the width of the 10 identified motifs ranged from 6–41 aa. Ten motifs were identified in the structure of the PgWRKYs, and their details are shown in Supplementary Figure 2. As shown in , motif 1 encoded the conserved WRKY domain, while motifs 2 and 3 encoded the conserved zinc-finger structure. Notably, similar motif compositions were observed in the same group of PgWRKY proteins. Most members within the same clade shared a unique motif composition compared to that in the other clades. For example, motif 1 and motif 2 were found in all 27 PgWRKY proteins, while motif 3 was not found in Group III members and was found in all other groups. Motif 7 exists only in two members of Group I and Group IIc (PgWRKY24 and PgWRKY26). Only members of Group I and Group II have motif 8. Motif 9 was found only in Group III. Generally, the closely-related PgWRKYs in the phylogenetic tree shared similar gene and motif compositions, suggesting that PgWRKYs in the same group may play similar functional roles.
Figure 2. Conserved motif analysis of PgWRKYs. The motif composition of PgWRKY was analyzed by the MEME tool. The detailed information of the ten motifs is in Supplementary Figure 2.
3.3 Phylogenetic analysis of PgWRKY
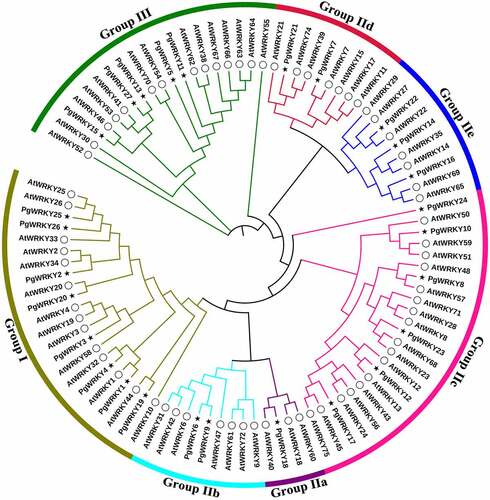
To investigate the evolution of PgWRKY family members, a phylogenetic tree was constructed using the maximum-likelihood estimate method using MEGA-X software and based on multiple alignments between full-length protein sequences of 72 AtWRKYs and 27 PgWRKYs. Phylogenetic analysis indicated that the classification of PgWRKY proteins was the same as that of A. thaliana, thereby confirming the classification accuracy of PgWRKY proteins. According to the constructed phylogenetic tree containing PgWRKYs and AtWRKYs (), PgWRKYs were classified into three primary groups (Groups I, II, and III). Among the 27 PgWRKY protein sequences, eight were assigned to Group I, fourteen to Group II, and five to Group III. Group II had the largest number of WRKY proteins and was divided into five major subgroups: one PgWRKY protein belonged to IIa (PgWRKY18), two to IIb (PgWRKY6 and PgWRKY9), six to IIc (PgWRKY8, PgWRKY10, PgWRKY12, PgWRKY17, PgWRKY23, and PgWRKY24), two to IId (PgWRKY7 and PgWRKY21) and, three to IIe (PgWRKY14, PgWRKY16, and PgWRKY22). Subgroups IIa and IIb were two subgroups in the same branch, whereas subgroups IId and IIe were derived from one clade. Notably, among these groups or subgroups, most members of the PgWRKYs were clustered in Group I. Overall, the classification of PgWRKYs confirms their diversification, which suggests that different family members may have varied functions.
Figure 3. Phylogenetic tree of the total WRKY sequences from P. grandiflorus and A. thaliana. The WRKY sequences were used for phylogenetic analysis using the MEGA-X software. The arcs with different colors represent seven subgroups of WRKY proteins. The solid black star and hollow circle represent WRKY sequences from P. grandiflorus and A. thaliana, respectively.
3.4 PgWRKYs expression profiles in different tissues of P. grandiflorus
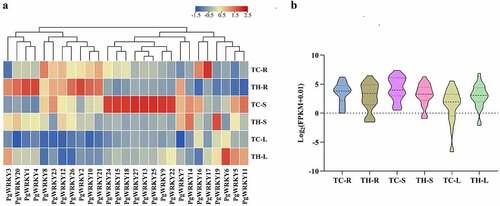
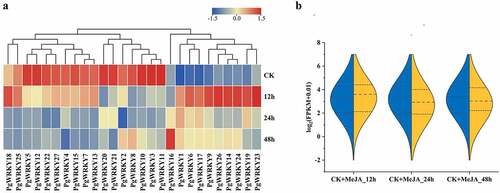
In this study, RNA-seq analysis was used to study the potential physiological functions of PgWRKY TFs ( and Supplementary Table 3). The expression levels of 27 genes in the roots, stems, and leaves of P. grandiflorus collected from Tongcheng southern Anhui (TC), and Taihe northern Anhui (TH), were investigated. Five genes were highly expressed in roots, and PgWRKY8 and PgWRKY17 in Group IIc were highly expressed in the roots of TC. PgWRKY1, PgWRKY2, and PgWRKY4 were highly expressed in the roots of TH, as were all members of Group I. Furthermore, PgWRKY6 was highly expressed in leaves, whereas eight genes (PgWRKY9, PgWRKY13, PgWRKY15, PgWRKY18, PgWRKY22, PgWRKY24, PgWRKY25, and PgWRKY27) were highly expressed in the stems. Based on the expression abundance of different groups, most members of Group III were highly expressed in all tissues (FPKM>5), and the genes in Groups I and II showed tissue-specific expression patterns, which might play an important role in the growth and development of P. grandiflorus. In addition, shows similar trends in the expression of roots from the two origins, differing in that the expression of PgWRKY genes was more concentrated in the roots from Tongcheng City. The expression trend of the PgWRKY gene in the stem of TC is higher than that in TH. In contrast, the PgWRKY gene showed an opposite trend with higher expression in the TH leaf compared to the TC. This indicates a difference in the expression level of PgWRKY genes in P. grandiflorus from northern and southern Anhui province.
Figure 4. Expression of PgWRKY genes in P. grandiflorus tissues. (A) The heatmap of all PgWRKY genes expression in different tissues. (B) The violin plot of all PgWRKY genes expression in different tissues.
3.5 Determination of triterpenoid saponins
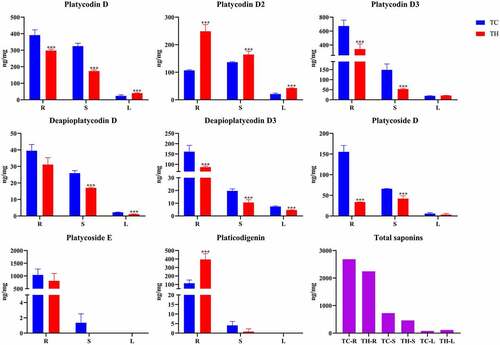
We used UHPLC-Orbitrap-MS/MS method to determine root, stem and leaf samples from Tongcheng City and Taihe County (3 replicates per organ). The contents of 8 saponins were expressed as mean and standard deviation (). In general, the content of triterpenoid saponins in roots was higher than that in stems and leaves. In different origins, the content of P. grandiflorus saponins also showed certain differences. The contents of Platycodin D, Platycodin D3, Deapioplatycodin D3 and Platycoside D in the roots and stems of TC were higher than those of TH. But the Platycodin D2 and Platicodigenin in TH Root is more than twice that of TC. Although the contents of Deapioplatycodin D and Deapioplatycodin D3 were lower in the two kinds of P. grandiflorus, the contents in TC stems and leaves were higher than those in TH. In addition, Platycodin D and Platycodin D2 were higher in TH leaves. Surprisingly, Platycoside E and Platicodigenin were not detected in the leaves from both origins. The total saponins content in roots and stems of TC was higher than that in TH, but the situation in leaves was opposite.
Figure 5. Saponin contents in different tissues of P. grandiflorus collected from Tongcheng southern Anhui (TC) and Taihe northern Anhui (TH) (mean ± SD, n = 3). ***Data compared with TC, P < 0.05.
3.6 Expression analysis of PgWRKY genes under MeJA treatment
The sequence and heat map of this experiment were based on the MeJA-treated transcriptome data (Supplementary Table 4). As shown in , the expression levels of most PgWRKYs treated with MeJA varied over time. The WRKY expression level was upregulated after 12 h, whereas it was downregulated at 24 and 48 h. Specifically, the transcription levels of nine genes (PgWRKY1, PgWRKY6, PgWRKY9, PgWRKY14, PgWRKY17, PgWRKY19, PgWRKY23, PgWRKY24 and PgWRKY26) increased rapidly under MeJA treatment, while those of the other 7 genes (PgWRKY2, PgWRKY3, PgWRKY8, PgWRKY10, PgWRKY11, PgWRKY20, and PgWRKY21) were lower under MeJA treatment than that in the control. In addition, the expression performance of some genes changed significantly in specific time frames. For example, the expression levels of PgWRKY2 and PgWRKY8 at 24 h were lower than those in the control but were significantly upregulated at 48 h.
Figure 6. Expression of PgWRKY genes in response to MeJA treatment. (A) The expression of PgWRKY genes for MeJA treatments. (B) Violin plot of PgWRKY genes before and after MeJA treatment.
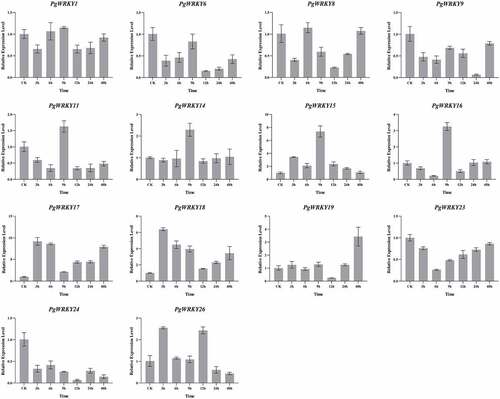
Fourteen PgWRKY genes were initially selected based on their expression levels in the RNA-seq data, and their performance was further validated by qRT-PCR analysis (). The expression trends of PgWRKY8, PgWRKY11, PgWRKY16, and PgWRKY26 were consistent with the transcriptome data. There were certain differences between the actual observations and transcriptome data, which can be explained by the individual differences between the material transcriptome sequencing and qRT-PCR analysis. Notably, the expression levels of PgWRKY17, PgWRKY18, and PgWRKY26 increased rapidly at 3 h. After 9 h of treatment, PgWRKY11, PgWRKY14, PgWRKY15, and PgWRKY16 also increased rapidly. From these data, it can be deduced that MeJA regulates the expression of these genes.
Figure 7. qRT-PCR analysis of PgWRKY genes under MeJA treatment. The Y- and X-axis represent the relative expression level and the time course of MeJA treatment, respectively. Roots were sampled at 0, 3, 6, 9, 12, 24, and 48 h after MeJA treatments. Data represent the mean ± SD of three technical repeats.
3.7 Co-expression analysis of candidate triterpenes saponin biosynthesis and PgWRKY genes
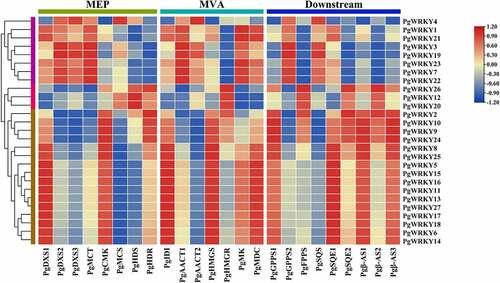
WRKY TFs are involved in various plant physiological activities. To elucidate the relationship between PgWRKYs and triterpenoid biosynthesis in P. grandiflorus, we constructed a co-expression network of PgWRKYs and genes related to the upstream 2-C-methyl-D-erythritol-4-phosphate (MEP) and mevalonate (MVA) pathways and downstream triterpene skeleton formation pathway ( and Supplementary Table 5). Co-expression analysis suggested that the correlation between PgWRKYs and genes related to the biosynthesis of triterpenoids in P. grandiflorus could be divided into three main clusters. In cluster I, several PgWRKY genes showed strong correlations with upstream genes in the MEP pathway, including PgDXS1, PgDXS2, PgDXS3, and PgMCT. The PgWRKYs in cluster I also showed strong correlations with MVA and some downstream genes, such as PgAACT1, PgMK, PgMDC, PgGPPS2, and PgSQS. In contrast, PgWRKYs in cluster II showed a low correlation with genes in the P. grandiflorus triterpenoid biosynthetic pathway and only correlated strongly with upstream PgHDS and PgHMGR. Some PgWRKYs in cluster III showed a strong positive correlation with pathway genes. PgDXS1 and PgCMK in the MEP pathway and PgIDI, PgHMGS, and PgMDC in the MVA pathway were strongly positively correlated with PgWRKYs in cluster III. Downstream, PgGPPS1, PgSQE1, Pgβ-AS1, and Pgβ-AS3 also showed a strong correlation with PgWRKYs in cluster III. PgWRKYs in cluster III may be involved in and regulate the biosynthesis of triterpenoids in P. grandiflorus. Co-expression analysis showed that MEP pathway genes were relevant to PgWRKYs in cluster I, and the MVA pathway and downstream pathway genes were relevant to PgWRKYs in cluster III. Four genes, PgWRKY2, PgWRKY9, PgWRKY10, and PgWRKY24, were selected for their modulatory potential in triterpene biosynthesis.
Figure 8. The Pearson’s correlation coefficients of PgWRKYs with triterpenoid biosynthesis pathway in P. grandiflorus. 1-Deoxy-d-xylulose-5-phosphate synthase (PgDXS); 2-C-methyl-d-erythritol 4-phosphate cytidylyltransferase (PgMCT); 4-diphosphocytidyl-2-C-methyl-D-erythritol kinase (PgCMK); 2-C-methyl-d-erythritol 2,4-cyclodiphosphate synthase (PgMCS); (E)-4-Hydroxy-3-methylbut-2-enyl-diphosphate synthase (PgHDS); 4-Hydroxy-3-methylbut-2-en-1-yl diphosphate reductase (PgHDR); Isopentenyl diphosphate isomerase (PgIDI); Acetyl-CoA C-acetyltransferase (PgAACT); Hydroxymethylglutaryl-CoA synthase (PgHMGS); Hydroxymethylglutaryl-CoA reductase (PgHMGR); Mevalonate kinase (PgMK); Diphosphomevalonate decarboxylase (PgMDC); Geranyl pyrophosphate synthase (PgGPPS); Farnesyl diphosphate synthase (PgFPPS); Squalene synthase (PgSQS); Squalene monooxygenase (PgSQE); β-Amyrin synthase (Pgβ-AS).
3.8 GO annotation and interaction analysis of specific PgWRKY proteins
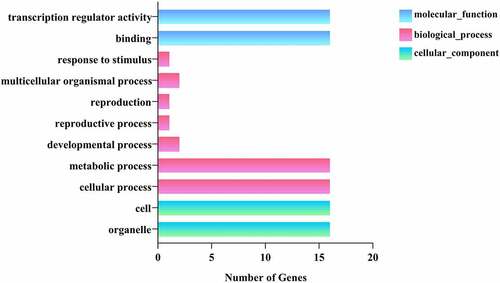
To further explore the biological and molecular functions of PgWRKY proteins, GO annotation and protein-protein interaction (PPI) network analysis were performed on the 27 PgWRKY proteins in this study. The GO annotation results consisted of biological processes (BP), cellular components (CC), and molecular functions (MF) (). Most PgWRKYs regulate cellular, metabolic, developmental, reproductive, and multicellular organismal processes, as well as responses to stimuli. The molecular functions of PgWRKYs are related to transcriptional regulatory activity and binding. Cellular components of this protein family include organelles and cells.
Figure 9. GO analysis of PgWRKY proteins. The results are grouped into three main categories: biological process, cellular component, and molecular function. The X-axis indicates the number of genes.
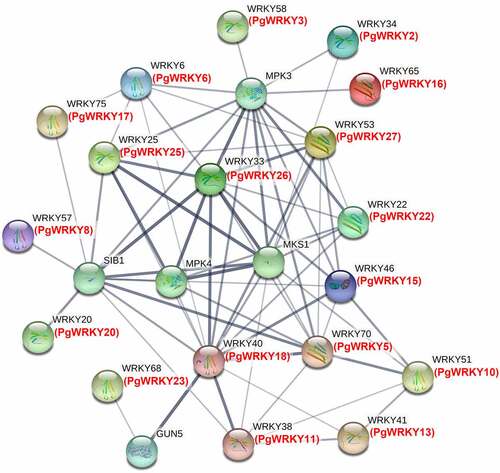
The STRING 11.5 online software was used to construct a WRKY protein-protein interaction network to discover the presence of PPIs. We identified five high confidence interacting proteins in the Arabidopsis WRKY family: VQ proteins (such as Meckel syndrome, type 1 [MKS1] and sigma factor binding protein 1 [SIB1]) involved in regulating plant defense responses, mitogen-activated protein kinase (MPK) 3 and MPK4 proteins involved in plant responses to pathogens and stresses, and genomes uncoupled 5 (GUN5) proteins involved in abiotic stress responses. As shown in , the PgWRKY33 protein is highly homologous to Arabidopsis WRKY33, suggesting that it may have potentially stronger interactions with most plant defense proteins (MPK3, MPK4, MKS1, and SIB1). Similarly, the PgWRKY18 protein is highly homologous to Arabidopsis WRKY40 and is presumed to have a strong interaction with the internal members of the WRKY family.
Figure 10. Protein-protein interaction network of specific PgWRKY proteins. Black and red color characters represent A. thaliana and P. grandiflorus, and the thick lines represent the strength of interaction.
4 Discussion
The WRKY gene family is one of the largest TF families that regulate plant growth and secondary metabolism.Citation18,Citation59 Highly advanced high-throughput sequencing technology and available public databases have facilitated the comprehensive study of several gene families.Citation60 The WRKY family has been studied in several model plants, cash crops, and medicinal plants such as Arabidopsis thaliana,Citation61 Sorghum bicolor,Citation62 Zea mays,Citation63 Panax ginseng,Citation29 Isatis tinctoria,Citation64 and Andrographis paniculata.Citation65 Our previous studies combined second- and third-generation sequencing technologies to systematically analyze the transcriptomes of different organs of P. grandiflorus.Citation49 Based on transcriptomic data in P. grandiflorus, 27 PgWRKYs were identified of P. grandiflorus for the first time. This study provides a reference for a further functional understanding of the WRKY gene family.
Most PgWRKY proteins contain a highly conserved heptapeptide at the N-terminus (WRKYGQK). However, at heptapeptide variant WRKYGKK was found in PgWRKY10. These differences may affect the ability of WRKY TFs to bind to the W-box. For example, soybean WRKY TFs with WRKYGKK do not properly bind to the W-box.Citation66 NtWRKY12 in Nicotiana tabacum has the WRKYGKK motif, and these transcription factors bind to the TTTTCCAC sequence but not to the W-box (TTGACT/C).Citation67 Therefore, we hypothesize that the heptapeptide variants may alter the binding properties of TFs to W-box and affect the transcriptional levels of PgWRKY transcription factors-targeted genes.
Based on sequence alignment and phylogenetic tree analysis ( and ), the WRKY family members were grouped similarly to Arabidopsis: Group I, Group II, and Group III, while Group II was further subdivided into subgroups IIa, IIb, IIc, IId, and IIe.Citation19 In this study, Group I had eight members, Group II had fourteen members, accounting for 51.85% of all PgWRKY genes, Group III had five members. These results are consistent with WRKY group sizes in Arabidopsis, maize, and ginseng.Citation19,Citation29,Citation63 Among these subgroups, IIc had the most members with six PgWRKY genes, accounting for 42.86% of the genes in Group II, which is similar to the results for soybean, Arabidopsis, and Andrographis.Citation19,Citation28,Citation65 Rinerson et al. Citation68 proposed four main lineages of WRKY transcription factor families in flowering plants: Group I + IIc, Group IIa + IIb, Group IId + IIe, and Group III, reflecting the evolution pathway of WRKY family members. This was also observed in the P. grandiflorus WRKY gene family. For example, members of Groups IIa and IIb were in the same evolutionary branch, and members from Groups IId and IIe were grouped into the same branch, suggesting that these subgroups arose indirectly from a common ancestor.
These motifs are associated with transcriptional activity and protein-protein interactions of target genes.Citation69 Therefore, the characterization and functions of TFs can be identified by analyzing conserved motif information. All PgWRKY genes had more than two conserved motifs, consistent with reports from other plant species.Citation70,Citation71 Each group member had the same or similar motif composition, suggesting that WRKY genes from the same group have similar protein structures and biological functions.
RNA-seq is an essential tool for gene function and structure studies at the global level to reveal the molecular mechanisms of specific biological processes. It has been widely used in basic research and drug development.Citation72,Citation73 We used transcriptome data form different tissues of P. grandiflorus from two origins to determine the WRKY family gene expression (). In the present study, two PgWRKYs of P. grandiflorus were two origins expressed differentially in roots, stems, and leaves. This indicated that PgWRKYs function in the growth and development of P. grandiflorus. All PgWRKY genes were expressed in at least one tissue (FPKM > 1), and their expression varied significantly among the different tissues. As shown in , PgWRKYs in roots and stems were higher than those in leaves, and the median FPKM values of root and stem tissues of Tongcheng City (south Anhui province) P. grandiflorus were higher than those of Taihe County (north Anhui province). This study also analyzed the differences in the content of 8 kinds of Platycodon saponins in different tissues of P. grandiflorus from the two origins mentioned above (). The highest saponin content was found in root tissues, followed by stems and leaves. The content of saponins in the roots and stems of Tongcheng P. grandiflorus is higher than that of Taihe. These are consistent with the expression trend of PgWRKY genes. Therefore, PgWRKYs may regulate the metabolic process of triterpenoids.
Notably, five genes showed the highest expression levels in roots. One gene was highly expressed in the leaves, whereas eight genes were highly expressed in the stems. These tissue-specific expression patterns suggest that PgWRKYs may be involved in tissue-specific developmental and signal transduction processes. For example, roots interact with the external environment, and genes highly expressed in the roots are more likely to participate in various stress response activities. AtWRKY39, AtWRKY48, and AtWRKY57 regulate plant heat tolerance, drought resistance, and immunity against pathogens.Citation74–76 PgWRKY8, PgWRKY17, and their orthologs in P. grandiflorus were specifically expressed in roots. They likely regulate plant growth and development by helping the underground plant parts defend against stress. Similarly, PgWRKY8, a highly expressed gene in the roots, and its homologous AaWRKY40 in A. annua are involved in the metabolism of terpenoids.Citation77 This indicates that PgWRKY8 may also regulate the metabolism of terpenoids in the roots. In addition, the wheat WRKY transcription factor gene TaWRKY71-1 was specifically expressed in leaves, and plants overexpressing TaWRKY71-1 had larger leaves and better growth ability than its parent common wheat JN177.Citation78 It is suggested that PgWRKY6 highly expressed in leaves has similar functions. Studying tissue-specific expression of PgWRKYs can provide critical information for further understanding the function of the P. grandiflorus WRKY gene family.
MeJA is involved in several physiological processes, such as plant growth and stress response. It regulates secondary metabolism in both angiosperms and gymnosperms.Citation79–81 Triterpene biosynthesis is regulated by MeJA signaling, and many MeJA-responsive TFs can regulate the MeJA signaling pathway.Citation14,Citation82 Therefore, WRKY genes in response to MeJA in P. grandiflorus may impact triterpenoid biosynthesis. Previous studies have shown that the accumulation of triterpenoid saponins in the hairy roots of P. grandiflorus increased after MeJA induction, and the transcription levels of triterpenoid biosynthesis pathway genes (PgHMGS, PgHMGR, PgMK, and PgMVD) increased rapidly at 3–6 h of MeJA treatment.Citation83 This study used publicly-available transcriptome data to study the PgWRKY genes expression under MeJA induction. A few genes (e.g., PgWRKY8, PgWRKY11, PgWRKY16, and PgWRKY26) showed the same expression trend as the RNA-seq analysis. But we found that the expression levels of some genes (PgWRKY15, PgWRKY17, PgWRKY18, and PgWRKY26) increased significantly at 3–6 h. This was consistent with the gene expression trend of triterpenoid biosynthesis pathway after MeJA treatment. It is speculated that PgWRKY15, PgWRKY17, PgWRKY18, and PgWRKY26 may indirectly regulate the biosynthesis of triterpenoid saponins by interacting with terpenoid synthase genes.
WRKY TFs are involved in plant development and regulate the biosynthesis of secondary metabolites. Medicinal plants with Terpenes are the main medicinal components of P. ginseng, A. apiacea, P. notoginseng, and P. quinquefolium. The terpenoid biosynthesis strategy in plants includes the MVA and MEP pathways.Citation84,Citation85 They are governed by multi-level network regulation, which can be accomplished by various structural genes in the biosynthetic pathway, and secondary regulation is accomplished by TFs.Citation86 P. ginseng transcription factors PgWRKY4X binds to the squalene epoxidase (PgSE) promoter to upregulate ginsenoside biosynthesis-related genes and highly improve the accumulation of ginsenosides.Citation87 Overexpression of PqWRKY1 activates the triterpenoid biosynthesis pathway in transformed Arabidopsis.Citation88 In this study, we found that four genes (PgWRKY2, PgWRKY9, PgWRKY10, and PgWRKY24) were positively correlated with triterpene biosynthesis pathway genes. These four genes were distributed in phylogenetic Groups I and II, which is in accordance with the related research results for P. ginseng.Citation29 The above co-expression results reflect the potential regulatory function of WRKYs in triterpenoid biosynthesis, and overexpression or knockout analyses of these PgWRKY genes will help clarify their functions.
GO annotation of gene families and the study of potential protein-protein interaction networks help understand the functions of these genes in this family.Citation89 GO analysis showed that the WRKY gene family of P. grandiflorus functions in three aspects: biological process, cellular component and molecular function. In A. thaliana, most studies on protein-protein interactions of WRKY TFs have focused on the AtWRKY33 protein involved in plant defense responses.Citation90 For example, Arabidopsis AtWRKY33 and AtWRKY25 interact with MKS1, a VQ protein that uses AtMPK4 as a substrate. Activated MPK4 phosphorylates MKS1 and releases AtWRKY33 after infection with the bacterial pathogen Pseudomonas syringae or flagellin. The released AtWRKY33 acts on the promoter of PAD3, which encodes a biosynthetic enzyme involved in producing phytoalexins.Citation91 In addition, dual-targeted SIB1 activates AtWRKY33 and functions in plant defenses against necrotrophic pathogens.Citation92 The protein-protein interaction network of P. grandiflorus showed that PgWRKY26 was highly homologous to AtWRKY33. These results suggest that PgWRKY26 may mediate interactions between different signaling pathways and hinge on the regulatory network of plant defense responses.
5 Conclusion
In total, 27 P. grandiflorus WRKY genes were identified in this study. The classification, evolutionary relationships, gene structure, and conserved motifs of the P. grandiflorus WRKY gene family were studied. The expression patterns of PgWRKY genes in different tissues of P. grandiflorus and their response to MeJA indicated that these genes might have essential functions in the growth, development, and secondary metabolism of P. grandiflorus. Co-expression analysis revealed a potential regulatory role of PgWRKYs in triterpenoid biosynthetic pathway genes. GO annotation and PPI analysis of PgWRKY proteins were performed to explore the functions and regulatory mechanisms of PgWRKY genes in P. grandiflorus. In conclusion, this study provides valuable information for further research on the regulatory mechanisms of WRKY TFs in plant growth, secondary metabolism, and resistance to various stressors.
6 Abbreviations
TFs, Transcription factors; P. grandifloras, Platycodon grandiflorus; MeJA, methyl jasmonate; MW, molecular weight; pI, isoelectric point; AA, amino acid; ORF, open reading frame; h, hours; qRT-PCR, quantitative real-time polymerase chain reaction; CK, control check; RNA-seq, RNA sequencing; MEME, multiple expectation maximizations for motif elicitation; RSEM, RNA-seq by expectation-maximization; GO, gene ontology; PPI, protein-protein interaction; BP, biological processes; CC, cellular components; MF, molecular functions; MKS1, Meckel syndrome, type 1; SIB1, sigma factor binding protein 1; MPK, mitogen-activated protein kinase; GUN5, genomes uncoupled 5; MVA, mevalonate; MEP, 2-C-methyl-D-erythritol-4-phosphate UHPLC-Orbitrap-MS/MS, ultra-high performance liquid chromatography-Orbitrap-tandem mass spectrometry.
Data availability
The raw data supporting the conclusions of this article will be made available by the authors without undue reservation to any qualified researcher.
Author contributions
LZ and SG conceived the study. BC and ND prepared and cultivated the plants. ML and JL prepared the samples, extracted RNA, and performed the qRT-PCR experiments. XC, JW, and HY assisted with data analysis and interpretation. JL and HY wrote the manuscript, and LZ revised the manuscript. All authors have reviewed, read, and approved the final manuscript.
Supplemental Material
Download Zip (4.2 MB)Acknowledgments
We would like to thank BGI Genomics for their assistance with the experiments.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15592324.2022.2089473
Additional information
Funding
Related Research Data
References
- Ji MY, Bo A, Yang M, Xu JF, Jiang LL, Zhou BC, Li, MH. The pharmacological effects and health benefits of Platycodon grandiflorus-a medicine food homology species. Foods. 2020;9(2): 142. doi:10.3390/foods9020142.
- Zhang LL, Huang MY, Yang Y, Huang MQ, Shi JJ, Zou L, Lu, JJ . Bioactive platycodins from platycodonis radix: phytochemistry, pharmacological activities, toxicology and pharmacokinetics. Food Chem. 2020;327:127029. doi:10.1016/j.foodchem.2020.127029.
- Zhang L, Wang Y, Yang D, Zhang C, Zhang N, Li M, Liu, Y. Platycodon grandiflorus - an ethnopharmacological, phytochemical and pharmacological review. J Ethnopharmacol. 2015;164:147–16. doi:10.1016/j.jep.2015.01.052.
- Jang K-J, Kim HK, Han MH, Oh YN, Yoon H-M, Chung YH, Kim, GY, Hwang, HJ, Kim, BW, Choi, YH. Anti-inflammatory effects of saponins derived from the roots of Platycodon grandiflorus in lipopolysaccharide-stimulated BV2 microglial cells. International journal of molecular medicine. 2013;31(6):199–206. doi:10.3892/ijmm.2013.1330.
- Buchwald W, Szulc M, Baraniak J, Derebecka N, Kania-Dobrowolska M, Piasecka A, Bogacz, A, Karasiewicz, M, Bartkowiak-Wieczorek, J, Kujawski, R et al, et al. The effect of different water extracts from Platycodon grandiflorum on selected factors associated with pathogenesis of chronic bronchitis in rats. Molecules. 2020;25(21):5020. doi: 10.3390/molecules25215020.
- Choi YH, Yoo DS, Cha MR, Choi CW, Kim YS, Choi SU, Lee, KR, Ryu, SY. Antiproliferative effects of saponins from the roots of Platycodon grandiflorum on cultured human tumor cells. Journal of natural products. 2010;73(11):1863–1867. doi:10.1021/np100496p.
- Hu Q, Pan R, Wang L, Peng B, Tang J, Liu X. Platycodon grandiflorum induces apoptosis in SKOV3 human ovarian cancer cells through mitochondrial-dependent pathway. Am J Chin Med. 2010;38(2):373–386. doi:10.1142/S0192415X10007919.
- Kim TW, Lee HK, Song IB, Lim JH, Cho ES, Son HY, Jung, JY, Yun, HI. Platycodin D attenuates bile duct ligation-induced hepatic injury and fibrosis in mice. Food Chem Toxicol. 2013;51:364–369. doi:10.1016/j.fct.2012.10.017.
- Fu CL, Liu Y, Leng J, Zhang J, He YF, Chen C, Wang, Z, Li, W. Platycodin D protects acetaminophen-induced hepatotoxicity by inhibiting hepatocyte MAPK pathway and apoptosis in C57BL/6J mice. Biomed Pharmacother. 2018;107:867–877. doi:10.1016/j.biopha.2018.08.082.
- Zhan X, Liao X, Luo X, Zhu Y, Feng S, Yu C, Lu, J, Shen, C, Wang, H. Comparative metabolomic and proteomic analyses reveal the regulation mechanism underlying meja-induced bioactive compound accumulation in cutleaf groundcherry (Physalis angulata L.) hairy roots. J Agric Food Chem. 2018;66(25):6336–6347. doi:10.1021/acs.jafc.8b02502.
- Bertini L, Palazzi L, Proietti S, Pollastri S, Arrigoni G, Polverino de Laureto P, Caruso, C . Proteomic analysis of Meja-induced defense responses in rice against wounding. Int J Mol Sci . 2019;20(10):2525. doi:10.3390/ijms20102525.
- Wang T, Zhang X. Genome-wide dynamic network analysis reveals the potential genes for MeJA-induced growth-to-defense transition. BMC plant biology. 2021;21(1):450. doi:10.1186/s12870-021-03185-1.
- Zheng XY, Lin TW, Du JF, Huang LJ, Li P, Lu X. A novel method for ginkgolide biosynthesis elucidation based on MeJA induction and differential metabolomics. J Chromatogr B Analyt Technol Biomed Life Sci. 2021;1176:122758. doi:10.1016/j.jchromb.2021.122758.
- Fu X, Peng B, Hassani D, Xie L, Liu H, Li Y, Chen T, Liu P, Tang Y, Li L, et al. AaWRKY9 contributes to light- and jasmonate-mediated to regulate the biosynthesis of artemisinin in Artemisia annua. New Phytol. 2021;231(5):1858–1874. doi:10.1111/nph.17453.
- Cui Q, Yan X, Gao X, Zhang DM, He HB, Jia GX. Analysis of WRKY transcription factors and characterization of two botrytis cinerea-responsive LrWRKY genes from lilium regale. Plant Physiol Biochem. 2018;127:525–536. doi:10.1016/j.plaphy.2018.04.027.
- Ji N, Li Y, Wang J, Zuo X, Li M, Jin P, Zheng, Y, et al. Interaction of PpWRKY46 and PpWRKY53 regulates energy metabolism in MeJA primed disease resistance of peach fruit. Plant Physiol Biochem. 2022;171:157–168. doi:10.1016/j.plaphy.2021.12.035.
- Ulker B, Somssich IE. WRKY transcription factors: from DNA binding towards biological function. Curr Opin Plant Biol. 2004;7(5):491–498. doi:10.1016/j.pbi.2004.07.012.
- Rushton PJ, Somssich IE, Ringler P, Shen QJ. WRKY transcription factors. Trends Plant Sci. 2010;15(5):247–258. doi:10.1016/j.tplants.2010.02.006.
- Eulgem T, Rushton PJ, Robatzek S, Somssich IE. The WRKY superfamily of plant transcription factors. Trends Plant Sci. 2000;5(5):199–206. doi:10.1016/S1360-1385(00)01600-9.
- Rushton PJ, Macdonald H, Huttly AK, Lazarus CM, Hooley R. Members of a new family of DNA-binding proteins bind to a conserved cis-element in the promoters of alpha-Amy2 genes. Plant Mol Biol. 1995;29(4):691–702. doi:10.1007/BF00041160.
- Zhang Y, Wang L. The WRKY transcription factor superfamily: its origin in eukaryotes and expansion in plants. BMC Evol Biol. 2005;5(1):1. doi:10.1186/1471-2148-5-1.
- Bakshi M, Oelmüller R. WRKY transcription factors: jack of many trades in plants. Plant Signal Behav. 2014;9(2):e27700. doi:10.4161/psb.27700.
- Gu L, Wang H, Wei H, Sun H, Li L, Chen P, Elasad M, Su Z, Zhang C, Ma L, et al. Identification, expression, and functional analysis of the group IId WRKY subfamily in upland cotton (Gossypium hirsutum L.). Front Plant Sci. 2018;9:1684. doi:10.3389/fpls.2018.01684.
- Gu L, Li L, Wei H, Wang H, Su J, Guo Y, Yu, S, et al. Identification of the group IIa WRKY subfamily and the functional analysis of GhWRKY17 in upland cotton (Gossypium hirsutum L.). PloS one. 2018;13(1):e0191681. doi:10.1371/journal.pone.0191681.
- Ishiguro S, Nakamura K. Characterization of a cDNA encoding a novel DNA-binding protein, SPF1, that recognizes SP8 sequences in the 5’ upstream regions of genes coding for sporamin and beta-amylase from sweet potato. Mol Geneti. 1994;244(6):563–571. doi:10.1007/BF00282746.
- Riechmann JL, Ratcliffe OJ. A genomic perspective on plant transcription factors. Curr Opin Plant Biol. 2000;3(5):423–434. doi:10.1016/S1369-5266(00)00107-2.
- Ross CA, Liu Y, Shen QJ. The WRKY gene family in rice (Oryza sativa). J Integr Plant Biol. 2007;49(6):827–842. doi:10.1111/j.1744-7909.2007.00504.x.
- Bencke-Malato M, Cabreira C, Wiebke-Strohm B, Bücker-Neto L, Mancini E, Osorio MB, Homrich MS, Turchetto-Zolet AC, De Carvalho MC, Stolf R, et al. Genome-wide annotation of the soybean WRKY family and functional characterization of genes involved in response to Phakopsora pachyrhiziinfection. BMC plant biology. 2014;14(1):236. doi:10.1186/s12870-014-0236-0.
- Di P, Wang P, Yan M, Han P, Huang X, Yin L, Yan Y, Xu Y, Wang Y. Genome-wide characterization and analysis of WRKY transcription factors in panax ginseng. BMC genomics. 2021;22(1):834. doi:10.1186/s12864-021-08145-5.
- Agarwal P, Reddy MP, Chikara J. WRKY: its structure, evolutionary relationship, DNA-binding selectivity, role in stress tolerance and development of plants. Mol Biol Rep. 2011;38(6):3883–3896. doi:10.1007/s11033-010-0504-5.
- Devaiah BN, Karthikeyan AS, Raghothama KG. WRKY75 transcription factor is a modulator of phosphate acquisition and root development in arabidopsis. Plant Physiol. 2007;143(4):1789–1801. doi:10.1104/pp.106.093971.
- Yang Y, Chi Y, Wang Z, Zhou Y, Fan B, Chen Z. Functional analysis of structurally related soybean GmWRKY58 and GmWRKY76 in plant growth and development. J Exp Bot. 2016;67(15):4727–4742. doi:10.1093/jxb/erw252.
- Cheng Y, Ahammed GJ, Yu J, Yao Z, Ruan M, Ye Q, Li Z, Wang R, Feng K, Zhou G, et al. Corrigendum: putative WRKYs associated with regulation of fruit ripening revealed by detailed expression analysis of the WRKY gene family in pepper. Sci Rep. 2017;7(1):43498. doi:10.1038/srep43498.
- Guan Y, Meng X, Khanna R, LaMontagne E, Liu Y, Zhang S. Phosphorylation of a WRKY transcription factor by MAPKs is required for pollen development and function in Arabidopsis. PLoS Genet. 2014;10(5):e1004384. doi:10.1371/journal.pgen.1004384.
- Jiang Y, Liang G, Yang S, Yu D. Arabidopsis WRKY57 functions as a node of convergence for jasmonic acid– and auxin-mediated signaling in jasmonic acid–induced leaf senescence. Plant Cell. 2014;26(1):230–245. doi:10.1105/tpc.113.117838.
- Miao Y, Zentgraf U. A HECT E3 ubiquitin ligase negatively regulates Arabidopsis leaf senescence through degradation of the transcription factor WRKY53. The Plant J: cell mol biol. 2010;63(2):179–188. doi:10.1111/j.1365-313X.2010.04233.x.
- Ma D, Pu G, Lei C, Ma L, Wang H, Guo Y, Chen J, Du Z, Wang H, Li G, et al. Isolation and characterization of AaWRKY1, an Artemisia annua transcription factor that regulates the amorpha-4,11-diene synthase gene, a key gene of artemisinin biosynthesis. Plant Cell Physiol. 2009;50(12):2146–2161. doi:10.1093/pcp/pcp149.
- AbuQamar S, Chen X, Dhawan R, Bluhm B, Salmeron J, Lam S, Dietrich, RA, Mengiste, T. Expression profiling and mutant analysis reveals complex regulatory networks involved in Arabidopsis response to botrytis infection. Plant J. 2006;48(1):28–44. doi:10.1111/j.1365-313X.2006.02849.x.
- Zheng Z, Qamar SA, Chen Z, Mengiste T. Arabidopsis WRKY33 transcription factor is required for resistance to necrotrophic fungal pathogens. The Plant J: cell mol biol. 2006;48(4):592–605. doi:10.1111/j.1365-313X.2006.02901.x.
- Qiao Z, Li CL, Zhang W. WRKY1 regulates stomatal movement in drought-stressed Arabidopsis thaliana. Plant Mol Biol. 2016;91(1–2):53–65. doi:10.1007/s11103-016-0441-3.
- Ren X, Chen Z, Liu Y, Zhang H, Zhang M, Liu Q, Hong, X, Zhu, JK, Gong, Z. ABO3, a WRKY transcription factor, mediates plant responses to abscisic acid and drought tolerance in arabidopsis. Plant J. 2010;63(3):417–429. doi:10.1111/j.1365-313X.2010.04248.x.
- Li S, Fu Q, Chen L, Huang W, Yu D. Arabidopsis thaliana WRKY25, WRKY26, and WRKY33 coordinate induction of plant thermotolerance. Planta. 2011;233(6):1237–1252. doi:10.1007/s00425-011-1375-2.
- Cheng Z, Luan Y, Meng J, Sun J, Tao J, Zhao D. WRKY transcription factor response to high-temperature stress. Plants. 2021;10(10). doi:10.3390/plants10102211.
- Yokotani N, Sato Y, Tanabe S, Chujo T, Shimizu T, Okada K, Yamane H, Shimono M, Sugano S, Takatsuji H, et al. WRKY76 is a rice transcriptional repressor playing opposite roles in blast disease resistance and cold stress tolerance. J Exp Bot. 2013;64(16):5085–5097. doi:10.1093/jxb/ert298.
- Tao Z, Kou Y, Liu H, Li X, Xiao J, Wang S. OsWRKY45 alleles play different roles in abscisic acid signalling and salt stress tolerance but similar roles in drought and cold tolerance in rice. J Exp Bot. 2011;62(14):4863–4874. doi:10.1093/jxb/err144.
- Lu H, Ju M, Chu S, Xu T, Huang Y, Chan Q, Peng, H, Gui, S . Quantitative and chemical fingerprint analysis for the quality evaluation of platycodi radix collected from various regions in China by HPLC coupled with chemometrics. Molecules . 2018;23(7):1823. doi:10.3390/molecules23071823 .
- Chang X, Li J, Ju M, Yu H, Zha L, Peng H, Wang, J, Peng, D, Gui, S. Untargeted metabolomics approach reveals the tissue-specific markers of balloon flower root (Platycodi Radix) using UPLC-Q-TOF/MS. Microchemical Journal. 2021;168:106447. doi:10.1016/j.microc.2021.106447.
- Yu J, Chang X, Peng H, Wang X, Wang J, Peng D, Gui, S. A strategy based on isocratic and linear-gradient high-speed counter-current chromatography for the comprehensive separation of platycosides from platycodi radix. Analytical methods: adv methods appl. 2021;13(4):477–483. doi:10.1039/D0AY02029J.
- Yu H, Liu M, Yin M, Shan T, Peng H, Wang J, Chang X, Peng D, Zha L, Gui S, et al. Transcriptome analysis identifies putative genes involved in triterpenoid biosynthesis in Platycodon grandiflorus. Planta. 2021;254(2):34. doi:10.1007/s00425-021-03677-2.
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. Clustal W and Clustal X version 2.0. Bioinf. 2007;23(21):2947–2948. doi:10.1093/bioinformatics/btm404.
- Bailey TL, Williams N, Misleh C, Li WW. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006;34:W369–73. doi:10.1093/nar/gkl198.
- Xie T, Chen C, Li C, Liu J, Liu C, He Y. Genome-wide investigation of WRKY gene family in pineapple: evolution and expression profiles during development and stress. BMC genomics. 2018;19(1):490. doi:10.1186/s12864-018-4880-x.
- Chen C, Chen H, Zhang Y, Thomas HR, Frank MH, He Y, Xia, R. TBtools: an integrative toolkit developed for interactive analyses of big biological data. Mol Plant. 2020;13(8):1194–1202. doi:10.1016/j.molp.2020.06.009.
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35(6):1547–1549. doi:10.1093/molbev/msy096.
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi:10.1038/nmeth.1923.
- Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011;12(1):323. doi:10.1186/1471-2105-12-323.
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. methods (San Diego. Calif). 2001;25:402–408 doi:10.1006/meth.2001.1262.
- Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, Doncheva NT, Legeay M, Fang T, Bork P, et al. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021;49(D1):D605–D612. doi:10.1093/nar/gkaa1074.
- Jiang J, Ma S, Ye N, Jiang M, Cao J, Zhang J. WRKY transcription factors in plant responses to stresses. J Integr Plant Biol. 2017;59(2):86–101. doi:10.1111/jipb.12513.
- Feller A, Machemer K, Braun EL, Grotewold E. Evolutionary and comparative analysis of MYB and bHLH plant transcription factors. The Plant J: cell mol biol. 2011;66(1):94–116. doi:10.1111/j.1365-313X.2010.04459.x.
- Dong J, Chen C, Chen Z. Expression profiles of the Arabidopsis WRKY gene superfamily during plant defense response. Plant Mol Biol. 2003;51(1):21–37. doi:10.1023/A:1020780022549.
- Baillo EH, Hanif MS, Guo Y, Zhang Z, Xu P, Algam SA. Genome-wide identification of WRKY transcription factor family members in sorghum (Sorghum bicolor (L.) moench). PloS one. 2020;15(8):e0236651. doi:10.1371/journal.pone.0236651.
- Hu W, Ren Q, Chen Y, Xu G, Qian Y. Genome-wide identification and analysis of WRKY gene family in maize provide insights into regulatory network in response to abiotic stresses. BMC plant biology. 2021;21(1):427. doi:10.1186/s12870-021-03206-z.
- Qu R, Cao Y, Tang X, Sun L, Wei L, Wang K. Identification and expression analysis of the WRKY gene family in Isatis indigotica. Gene. 2021;783:145561. doi:10.1016/j.gene.2021.145561.
- Zhang R, Chen Z, Zhang L, Yao W, Xu Z, Liao B, Mi, Y, Gao, H, Jiang, C, Duan, L, et al. Genomic characterization of wrky transcription factors related to andrographolide biosynthesis in andrographis paniculata. Frontiers in genetics. 2020;11:601689. doi:10.3389/fgene.2020.601689.
- Bi C, Xu Y, Ye Q, Yin T, Ye N. Genome-wide identification and characterization of WRKY gene family in Salix suchowensis. PeerJ. 2016;4:e2437. doi:10.7717/peerj.2437.
- van Verk Mc, Pappaioannou D, Neeleman L, Bol JF, Linthorst HJ. A novel wrky transcription factor is required for induction of PR-1a gene expression by salicylic acid and bacterial elicitors. Plant Physiol. 2008;146(4):1983–1995. doi:10.1104/pp.107.112789.
- Rinerson CI, Rabara RC, Tripathi P, Shen QJ, Rushton PJ. The evolution of WRKY transcription factors. BMC plant biology. 2015;15(1):66. doi:10.1186/s12870-015-0456-y.
- Liu L, White MJ, MacRae TH. Transcription factors and their genes in higher plants functional domains, evolution and regulation. Eur J biochem. 1999;262(2):247–257. doi:10.1046/j.1432-1327.1999.00349.x.
- Wei Y, Shi H, Xia Z, Tie W, Ding Z, Yan Y, Wang, W, Hu, W, Li, K. Genome-Wide identification and expression analysis of the WRKY gene family in cassava. Front Plant Sci. 2016;7:25. doi:10.3389/fpls.2016.00025.
- Xu YH, Sun PW, Tang XL, Gao ZH, Zhang Z, Wei JH. Genome-wide analysis of WRKY transcription factors in Aquilaria sinensis (Lour.) Gilg. Sci Rep. 2020;10(1):3018. doi:10.1038/s41598-020-59597-w.
- Xanthopoulou A, Ganopoulos I, Psomopoulos F, Manioudaki M, Moysiadis T, Kapazoglou A, Osathanunkul M, Michailidou S, Kalivas A, Tsaftaris A, et al. De novo comparative transcriptome analysis of genes involved in fruit morphology of pumpkin cultivars with extreme size difference and development of EST-SSR markers. Gene. 2017;622:50–66. doi:10.1016/j.gene.2017.04.035.
- Zhang H, Li M, Kong M, Dunwell JM, Zhang Y, Yue C, Wu, J, Zhang, S. Study on the differences of gene expression between pear and apple wild cultivation materials based on RNA-seq technique. BMC plant biology. 2021;21(1):256. doi:10.1186/s12870-021-03051-0.
- Xing DH, Lai ZB, Zheng ZY, Vinod KM, Fan BF, Chen ZX. Stress- and pathogen-induced Arabidopsis WRKY48 is a transcriptional activator that represses plant basal defense. Mol Plant. 2008;1(3):459–470. doi:10.1093/mp/ssn020.
- Jiang Y, Qiu Y, Hu Y, Yu D. Heterologous expression of AtWRKY57 confers drought tolerance in Oryza sativa. Front Plant Sci. 2016;7:145. doi:10.3389/fpls.2016.00145.
- Li S, Zhou X, Chen L, Huang W, Yu D. Functional characterization of Arabidopsis thaliana WRKY39 in heat stress. Mol Cells. 2010;29(5):475–483. doi:10.1007/s10059-010-0059-2.
- De Paolis A, Caretto S, Quarta A, Di Sansebastiano GP, Sbrocca I, Mita G, Frugis, G . Genome-Wide identification of wrky genes in Artemisia annua: characterization of a putative ortholog of AtWRKY40. Plants. 2020;9(12):1669. doi:10.3390/plants9121669.
- Qin Z, Lv H, Zhu X, Meng C, Quan T, Wang M, Xia G. Ectopic expression of a wheat WRKY transcription factor gene TaWRKY71-1 results in hyponastic leaves in Arabidopsis thaliana. PloS one. 2013;8(5):e63033. doi:10.1371/journal.pone.0063033.
- Wasternack C. Jasmonates: an update on biosynthesis, signal transduction and action in plant stress response, growth and development. Ann Bot. 2007;100(4):681–697. doi:10.1093/aob/mcm079.
- Pauwels L, Inzé D, Goossens A. Jasmonate-inducible gene: what does it mean? Trends Plant Sci. 2009;14(2):87–91. doi:10.1016/j.tplants.2008.11.005.
- Browse J, Pauwels L, Inzé D, Goossens A. Jasmonate passes muster: a receptor and targets for the defense hormone. Annu Rev Plant Biol. 2009;60(1):183–205. doi:10.1146/annurev.arplant.043008.092007.
- Wu Z, Li L, Liu H, Yan X, Ma Y, Li Y, Chen, T, Wang, C, Xie, L, Hao, X, et al. AaMYB15, an R2R3-MYB TF in Artemisia annua, acts as a negative regulator of artemisinin biosynthesis. Plant science: Int J Exp plant bio. 2021;308:110920. doi:10.1016/j.plantsci.2021.110920.
- Kim YK, Sathasivam R, Kim YB, Kim JK, Park SU. Transcriptomic analysis, cloning, characterization, and expression analysis of triterpene biosynthetic genes and triterpene accumulation in the hairy roots of Platycodon grandiflorum exposed to methyl jasmonate. ACS omega. 2021;6(19):12820–12830. doi:10.1021/acsomega.1c01202.
- Banerjee A, Sharkey TD. Methylerythritol 4-phosphate (MEP) pathway metabolic regulation. Natural product reports. 2014;31(8):1043–1055. doi:10.1039/C3NP70124G.
- Bach TJ, Boronat A, Caelles C, Ferrer A, Weber T, Wettstein A. Aspects related to mevalonate biosynthesis in plants. Lipids. 1991;26(8):637–648. doi:10.1007/BF02536429.
- Xu Y, Zhu C, Xu C, Sun J, Grierson D, Zhang B, Chen, K . Integration of metabolite profiling and transcriptome analysis reveals genes related to volatile terpenoid metabolism in finger citron (C. medica var. sarcodactylis). Molecules . 2019;24(14):2564. doi:10.3390/molecules24142564.
- Yao L, Wang J, Sun J, He J, Paek K-Y, Park S-Y, Huang, L, Gao, W. A WRKY transcription factor, PgWRKY4X, positively regulates ginsenoside biosynthesis by activating squalene epoxidase transcription in panax ginseng. Industrial Crops and Products. 2020;154:112671. doi:10.1016/j.indcrop.2020.112671.
- Sun Y, Niu Y, Xu J, Li Y, Luo H, Zhu Y, Liu M, Wu Q, Song J, Sun C, et al. Discovery of WRKY transcription factors through transcriptome analysis and characterization of a novel methyl jasmonate-inducible PqWRKY1 gene from panax quinquefolius. Plant Cell, Tissue and Organ Culture (PCTOC). 2013;114(2):269–277. doi:10.1007/s11240-013-0323-1.
- Tohge T, Fernie AR. Combining genetic diversity, informatics and metabolomics to facilitate annotation of plant gene function. Nat Protoc. 2010;5(6):1210–1227. doi:10.1038/nprot.2010.82.
- Chi Y, Yang Y, Zhou Y, Zhou J, Fan B, Yu JQ, Chen, Z. Protein-protein interactions in the regulation of WRKY transcription factors. Mol Plant. 2013;6(2):287–300. doi:10.1093/mp/sst026.
- Qiu JL, Fiil BK, Petersen K, Nielsen HB, Botanga CJ, Thorgrimsen S, Palma K, Suarez-Rodriguez MC, Sandbech-Clausen S, Lichota J, et al. Arabidopsis MAP kinase 4 regulates gene expression through transcription factor release in the nucleus. EMBO J. 2008;27(16):2214–2221. doi:10.1038/emboj.2008.147.
- Lai Z, Li Y, Wang F, Cheng Y, Fan B, Yu JQ, Chen, Z. Arabidopsis Sigma factor binding proteins are activators of the WRKY33 transcription factor in plant defense. Plant Cell. 2011;23(10):3824–3841. doi:10.1105/tpc.111.090571.