
Abstract
Objectives: Anxiety and adjustment disorders are among the most prevalent mental health conditions. This review focuses on γ-aminobutyric acid receptor type A (GABAAR)-mediated anxiolysis, describing the action of both endogenous and exogenous modulators of GABAAR. Future directions and innovative strategies to alleviate anxiety symptoms are discussed, with a particular emphasis on etifoxine.
Methods: We used available data from the recent literature to update the mode of action of anxiolytics. We focussed our search on anxiolytics acting at GABAARs, as well as on the pharmacological properties of formerly and currently prescribed anxiolytics.
Results: Considering the adverse effects of current treatments aimed at increasing inhibitory controls, optimisation of existing pharmacotherapies is of crucial importance. Among the alternative compounds targeting the GABAergic system, translocator protein (TSPO) ligands, such as etifoxine (EFX), which promote endogenous neurosteroidogenesis, are emerging as promising candidates for anxiety relief. In several papers comparing the efficacy of benzodiazepines and EFX, EFX showed interesting properties with limited side effects. Indeed, neurosteroids are potent GABAAR modulators with highly underrated anxiolytic properties.
Conclusions: Novel therapeutic strategies have been emerging following the recognition of neurosteroids as potent anxiolytics. Featured at the top of the list for well-tolerated anxiety relief, TSPO ligands such as etifoxine appear promising.
1. Introduction
Anxiety is an emotional experience that is characterised by a state of arousal and the expectation of danger (Gutierrez-Garcia and Contreras Citation2013). It has an adaptive role, preparing the body to deal with future threatening stimuli arising from the environment. Nevertheless, processing of such aversive information by the brain, particularly by the amygdala circuits, is associated with a negative subjective state. When excessive, then a pathological anxious condition may be diagnosed. In DSM-5, such states include panic disorder, generalised anxiety disorder, separation anxiety disorder, specific phobias, social anxiety disorder and adjustment disorder with anxiety.
Anxiety and adjustment disorders are excessively prevalent worldwide, and frequently comorbid with other medical conditions. Although the neurobiological mechanisms underlying anxiety are still poorly understood, treatment of anxiety conditions has largely improved over the last decades. In the first half of the twentieth century, barbiturates were widely used for their sedative properties, but major drawbacks, such as dependence and lethal overdose, led to their progressive replacement by benzodiazepines (Johns Citation1977). Recent investigation has led to the discovery of new circuits involved in anxiety, thus broadening potential treatment strategies and giving rise to new therapeutic opportunities (Calhoon and Tye Citation2015). Optimisation of treatment is of crucial importance, considering the substantial side effects of current anxiolytics.
Since anxious states are classically associated with neuronal hyperexcitability in structures processing emotions, the first line of treatment was thus aimed at increasing inhibitory control, mainly by targeting γ-aminobutyric acid receptor type A (GABAAR) in the central nervous system. In this paper, we review the molecular pharmacology of GABAAR and its most potent modulators, with a particular emphasis on etifoxine, an interesting anxiolytic with double-targeting properties.
2. Molecular pharmacology of GABAARs
GABAAR is a heteropentamer made of five subunits delimiting a chloride/bicarbonate-permeable channel (Olsen Citation2018). It is activated by at least two molecules of γ-aminobutyric acid (GABA), an amino acid neurotransmitter synthesised from glutamate in neurons expressing glutamate decarboxylase enzymes (i.e. GABAergic neurons). As for many ligand-gated receptor-channels the apparent affinity of GABAARs for GABA varies from low to high micromolar range. This parameter strongly depends on the subunit composition and, more specifically, on α-β subunit dimers forming together the agonist binding site for GABA.
Although about 19 subunits have been identified (), there is a limited set of receptors identified in the whole nervous system. Molecular genetics and pharmacological experiments suggest the actual existence of less than 10 different GABAAR subtypes, which is legitimate since two αβ dimers per receptor are at least required to activate GABAAR-channels. Among the identified native GABAAR-channels (Olsen and Sieghart Citation2009), those composed of α1β2γ2 are presumed to account for about 60% of all GABAARs, while minor forms are likely to be constituted by the following subunit combinations: α2β3γ2 (15–20%), α3βXγ2 (10–15%), α4βXγX or α4βXδ (5%), α5β2γ2 (less than 5%), α6β2/3γ2 (less than 5%) (Mohler et al. Citation2002).
Figure 1. Schematic representation of GABAA receptors (GABAARs) as heteropentameric complexes, made up of a combination of 19 subunits, delimiting a chloride-permeable channel when activated by at least two molecules of GABA. Extracellular sites for benzodiazepines and etifoxine, two anxiolytics, are indicated on the top view representation of the receptor channel, composed in the vast majority by αβγ subunits.
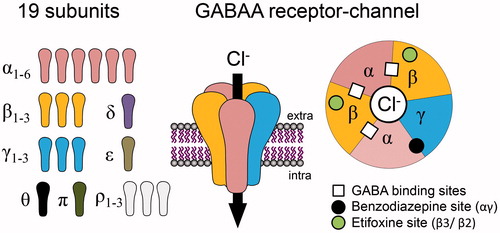
GABAAR activation is associated with an increase in chloride permeability through the plasma membrane, which is responsible for neuronal inhibition. This inhibition is generally seen as a membrane hyperpolarisation but, alternatively, may also consist of a shunting inhibition due to a local drop in membrane resistance. Both mechanisms reduce the capacity of neurons to produce action potentials or to regenerate these along their progression to synaptic endings. Recent findings have highlighted the importance of chloride gradients and their plasticity in physiological and pathological conditions. Indeed, destruction of these chloride gradients seems to contribute to several hyperexcitable neuropathologies such as epilepsy or neuropathic pain (Coull et al. Citation2003; Sammler et al. Citation2015). As illustrated in and , a large variety of compounds can bind to GABAARs. This includes endogenous modulators (e.g. endozepines and neuroactive steroids such as allopregnanolone) as well as anxiolytics such as benzodiazepines (e.g. diazepam, lorazepam), barbiturates or etifoxine.
Table 1. List of some ligands exerting a positive modulation on GABAAR function. Binding sites, if identified, are indicated in the table with their specific properties. In the case of steroids, access to the binding site is presumed to be intracellular or via membrane lateral mobility. All other compounds bind to extracellular binding sites.
3. Endogenous modulators of GABAAR function with anxiolytic properties
3.1. Endozepines
A diversity of endogenous compounds, including endozepines, can modulate GABAAR activity. Endozepines bind to the benzodiazepine site of GABAAR, as demonstrated by their ability to displace diazepam from its binding site (Costa and Guidotti Citation1991). DBI (diazepam binding inhibitor, recently renamed ACBD1-acyl-coenzyme A binding domain containing 1 protein (Fan et al. Citation2010)) and its two major bioactive cleavage products, TTN (triakontatetraneuropeptide) and ODN (octadecaneuropeptide), are the main endozepines. Their physiological role is controversial, with the majority of research suggesting that endozepines are negative allosteric modulators (Guidotti et al. Citation1983; Bormann Citation1991), but some authors have accumulated evidence implying otherwise (Christian et al. Citation2013). Although little is known about endozepines, astrocytes seem to play a major role in the release of DBI and its conversion into active peptides (Loomis et al. Citation2010). Endozepines also bind to a mitochondrial translocator protein (TSPO), formerly known as the peripheral benzodiazepine receptor, which will be detailed more thoroughly later in this review.
3.2. Neuroactive steroids
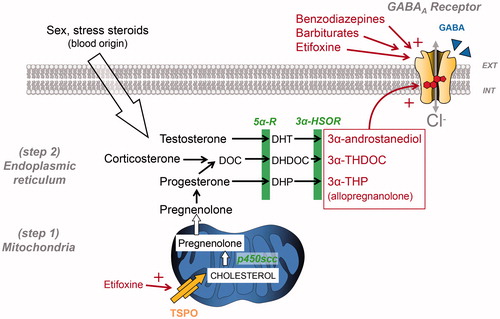
Neuroactive steroids have long been known to produce GABA-mediated anxiolysis (Longone et al. Citation2011; Schule et al. Citation2011); however, their precise mechanism of action is still being investigated. More precisely, 3α-reduced metabolites of progesterone (allopregnanolone), testosterone (3α-androstanediol) and corticosterone (tetrahydrodeoxycorticosterone), are recognised as the most potent endogenous GABAAR-positive allosteric modulators. When present at high nanomolar concentrations, they increase the opening burst duration of GABAAR chloride channels as well as the mean open time (Callachan et al. Citation1987; Twyman and Macdonald Citation1992). Synthesis of 3α-reduced neurosteroids starts with the stimulation of cholesterol transport into the mitochondria through an 18-kDa translocator protein (TSPO), expressed on the outer mitochondrial membrane (, see also (Rupprecht et al. Citation2010)). Interestingly, TSPO was previously called ‘peripheral benzodiazepine receptor’ since TSPO protein complexes possess binding sites for benzodiazepines as well as binding sites for other endogenous (e.g. protoporphyrin IX) and exogenous substances (e.g. etifoxine). The molecular action of these substances stimulates neurosteroidogenesis and the production of anxiolytic neurosteroids (Wolf et al. Citation2015). It is important to mention here that some metabolites, namely sulphated steroids and 3β-hydroxysteroids, may also have a negative modulatory role, sometimes as antagonist at neurosteroid binding sites or as allosteric negative modulators (Wang et al. Citation2002; Chisari et al. Citation2010).
Figure 2. Simplified view of neurosteroidogenesis focussing on the synthesis of 3α-reduced neurosteroid anxiolytics targeting GABAARs. This production is fully ensured by nerve cells if cholesterol is used as a precursor or can be made possible by using circulating sex and stress steroids, if they reach nerve cells. Note that 3α-reduced neurosteroids are likely to reach their GABAAR binding sites by lateral membrane diffusion. Cholesterol transport by translocator protein (TSPO) has been shown to be stimulated by several compounds including etifoxine, which also exerts an allosteric modulation of GABAARs. DHDOC: dehydro-deoxycorticosterone; DHP: dihydroprogesterone; DHT: dihydrotestosterone; DOC: deoxycorticosterone; THDOC: tetrahydrodeoxycorticosterone; p450scc, cytochrome p450 enzyme side chain cleavage; 5αR, 5α-reductase; 3α-HSOR, 3α-hydroxysteroid dehydrogenase.
Recent work has demonstrated the existence of two binding sites for neurosteroids on GABAAR, distinct from those of other modulatory compounds. For instance, neurosteroids modulate GABA activity by binding to α-subunit M1 and M4 transmembrane segments, whereas direct activation of the receptor requires binding to the interface between α-subunit M1 and β-subunit M3 (Hosie et al. Citation2006). Since neurosteroids are lipophilic molecules, they are likely reaching their site of action through membrane lateral diffusion (Akk et al. Citation2005). The strong affinity of neurosteroids for extrasynaptic δ-expressing GABAAR is also worth mentioning, considering their strong influence on neuronal excitability (Stell et al. Citation2003). At concentrations above 100 nM, neurosteroids develop GABA mimetic properties and can directly activate the receptor (Shu et al. Citation2004).
Besides their rapid effects on membrane receptors, neurosteroids also are part of the broader steroid group, and as such possess a genomic effect, impacting the transcriptional activity of numerous genes of interest. For example, allopregnanolone has been shown to inhibit corticotropin-releasing hormone (CRH) gene expression, therefore limiting CRH-induced anxiety (Patchev et al. Citation1994). It is also worth mentioning that sex steroids such as progesterone and oestradiol also have an impact on anxiety, through action in the amygdala (Frye and Walf Citation2004). Apart from the direct modulation of GABAAR function, increase in receptor expression mediated by sex hormone genomic modulation may also account for the anxiolytic effect of neurosteroids, as shown for α4 and δ subunits (Carver and Reddy Citation2016).
4. Pharmacological properties of prescribed anxiolytics acting at GABAARs
4.1. Barbiturates
Besides facilitating chloride channel opening, barbiturates also increase opening duration, and can act as GABA mimetics at high concentrations (ffrench-Mullen et al. Citation1993). Therefore, their inhibitory potential is much greater than that of benzodiazepines, which accounts for a higher risk of depression of the central nervous system. Exact binding sites of barbiturates on GABAAR have yet to be identified, although recent studies suggest the importance of the α/β and γ/β subunit interfaces (Chiara et al. Citation2013; Olsen Citation2018). Barbiturates can be classified according to their pharmacokinetic properties, with the intermediate-acting derivatives being used as anxiolytics. Despite the discontinuation of their use for anxiety disorders, barbiturates are still indicated in various conditions, such as seizure disorder and preoperative sedation.
4.2. Benzodiazepines
Benzodiazepines are one of the most used groups of prescription drugs around the world. They produce a potent anxiolysis but also possess sedative, muscle relaxant and anticonvulsant properties. Unfortunately, their use is impaired by major unwanted side effects such as retrograde amnesia, functional tolerance and a high dependence risk. In hippocampal neurons, it has been shown, for example, that withdrawal from a 5-day treatment with flunitrazepam in adult rats was sufficient to silence GABAergic synapses and produce severe withdrawal symptoms (Poisbeau, Williams et al. Citation1997). These side effects are particularly hard to manage in the long term, and this is critical in the elderly since benzodiazepines may also lead to psychomotor impairment and promote cognitive decline.
Benzodiazepine sites are found at the αγ subunit interface, and benzodiazepine affinity depends on α subunit identity (Olsen Citation2018). For example, α1 subunit-containing receptors are highly sensitive to most benzodiazepines (and to zolpidem) whereas those containing α2 or α3 are less sensitive to benzodiazepines. If benzodiazepines act as positive allosteric modulators, i.e. potentiating GABAAR function by increasing the affinity for GABA, modulation at this αγ site can be blocked by the antagonist flumazenil (also called a silent agonist) or negatively modulated by the β-carbolines.
The recent development of α subunit-selective benzodiazepines with possible limited adverse side effects showed that targeting the α2-containing GABAARs was the most efficient way to promote anxiolysis but also analgesia and sedation (). These studies also showed that potentiating α1-containing GABAARs could lead to major sedative and amnesic effects, while cognitive decline could be mostly attributed to the potentiation of α5-containing GABAARs. This opens a new avenue for the development of symptom-specific benzodiazepines but, so far, refinement of benzodiazepine molecules has not proven its efficacy to prevent functional tolerance or benzodiazepine dependence upon cessation of their use.
Table 2. Summary of the effects of benzodiazepines, either clinically relevant or adverse side effects, and the presumed α subunit composition of GABAARs (Adapted from (Tan et al. Citation2011)). Note that α4 or α6 subunit-containing GABAARs containing are not shown since they are benzodiazepine insensitive.
Ligand binding at αγ benzodiazepine sites on GABAARs may rapidly be associated with an uncoupling between the GABA and benzodiazepine site, likely explaining its loss of function (i.e. functional tolerance). Apart from this acute tolerance explanation, long-term use of benzodiazepine is associated with changes in the membrane expression of GABAARs. These newly expressed receptors often display a lower affinity to benzodiazepines or even a full pharmacoresistance, as seen sometimes in temporal lobe epilepsy (Leroy et al. Citation2004). The molecular mechanisms explaining these changes are still poorly understood but likely involve intracellular phosphorylation processes, as suggested by in vitro studies (Ali and Olsen Citation2001). This intracellular pathway seems to be strongly recruited when benzodiazepines promote sedation, i.e. after activation of α1γ-containing GABAARs. Taken together, it is very clear that high-affinity benzodiazepine binding sites contribute to a large part to functional tolerance and the main challenge to treat anxiety states is to design new benzodiazepine compounds that target α2γ-containing receptors.
An alternative to benzodiazepines is to use non-benzodiazepine compounds with distinct binding sites on GABAARs and limited adverse side effects. Etifoxine (Stresam) is one of the rare anxiolytics that is available on the market and that displays this property.
5. Etifoxine, a non-benzodiazepine anxiolytic
Etifoxine is a non-benzodiazepine (benzoxazine-derived) anxiolytic prescribed in several countries, including in France since 1979, for the treatment of adjustment disorders with anxiety (Servant et al. Citation1998; Nguyen et al. Citation2006). Its anxiolytic effect involves potentiation of GABAAR function, but etifoxine’s mechanism of action is quite original as it seems to use both direct and indirect pathways.
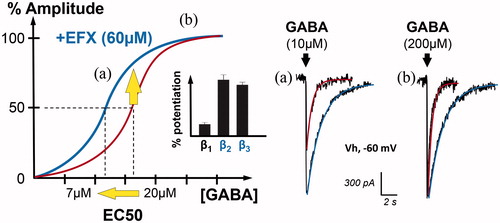
The direct positive allosteric modulation is observed after binding to a selective site, distinct from that of benzodiazepines (Verleye et al. Citation1999). Indeed, etifoxine binds to GABAARs on β subunits (Hamon et al. Citation2003), with a marked preference for β2 and β3 ( and ). As illustrated in , this binding leads to a positive allosteric modulation of chloride currents resulting from the opening of GABAARs (Schlichter et al. Citation2000). Increase in the affinity for the GABA binding site results in an overall decrease of the half maximal amplitude (EC50) (i.e. concentration of GABA producing a 50% modulation in the dose–response curve), an increase in the current amplitude in the case of non-saturating GABA concentrations and an increase in the current duration (see examples in ).
Figure 3. Positive allosteric modulation of GABAAR currents by the anxiolytic etifoxine (EFX). Graph on the left illustrates the GABAAR current amplitude (in %) when spinal neurons are submitted to increasing concentrations of GABA (red). In the presence of etifoxine (EFX, 60 µM), the dose–response curve is shifted to the left, indicating an increase in the apparent affinity for GABA. Indeed, the concentration of GABA inducing GABAAR-mediated current of half the maximal amplitude (EC50) is reduced from 20 to 7 µM. Bar chart in inset indicates that this modulation is more selective for GABAAR containing β2 and β3 subunits (adapted from (Hamon et al. Citation2003)). Another representation of this effect is shown on the right with representative traces. Etifoxine increased the amplitude and duration of GABAAR-mediated currents evoked by non-saturating concentration of GABA (a, 10 µM). In the case of saturating concentration (b, 200 µM), EFX only increased the duration of the current, as predicted by the dose–response curve.
As shown in , etifoxine also binds to the mitochondrial TSPO. This specific protein has recently received some attention since it may be a promising target for the development of novel anxiolytics with minimal adverse side effects (Nothdurfter et al. Citation2012). Binding of etifoxine to TSPO favours the entry of cholesterol into the mitochondria and its conversion by several enzymes to the potent endogenous anxiolytic called allopregnanolone (Verleye et al. Citation2005; do Rego et al. Citation2015; Wolf et al. Citation2015). Anxiolysis is then achieved by a neurosteroid-based potentiation of GABAAR function. Together, the direct and indirect actions of etifoxine on GABAARs lead to synergistic and long-lasting anxiolytic effects, at least in animal models (Verleye and Gillardin Citation2004; Ugale et al. Citation2007; Verleye et al. Citation2009, Citation2011).
The potentiation of GABAAR function is not exclusively restricted to a global anxiolytic action of etifoxine. Besides its initial description as an anticonvulsant (Kruse and Kuch Citation1985, Citation1986), etifoxine’s preclinical efficacy has been proven to limit acute pain expression (Juif et al. Citation2015), to alleviate pain symptoms resulting from an inflammatory or neuropathic insult (Aouad et al. Citation2009; Aouad, Petit-Demouliere, et al. Citation2014; Aouad, Zell, et al. Citation2014; Zeitler et al. Citation2016; reviewed in Poisbeau et al. Citation2014; Choi and Kim Citation2015), to exert a neuroprotective action in various models (Girard P et al. Citation2009; Ravikumar et al. Citation2016; Simon-O'Brien et al. Citation2016) and to promote nerve regeneration (Girard C et al. Citation2008; Zhou et al. Citation2013, Citation2014). As a follow-up of several preclinical reports from our laboratory (Aouad et al. Citation2009; Aouad, Petit-Demouliere, et al. Citation2014; Aouad, Zell, et al. Citation2014; Poisbeau et al. Citation2014; Juif et al. Citation2015; Zeitler et al. Citation2016) and others, a recent review has also highlighted the potential interest of using etifoxine for pain patients with anxiety (Choi and Kim Citation2015).
6. Future directions
Considering the risks associated with benzodiazepines, tremendous effort has been devoted to the development of novel anxiolytic strategies. These include compounds specifically targeting the α2 and α3 GABAAR subunits, which are the most relevant for anxiolysis. Although promising results were found in preclinical research (Griebel et al. Citation2003), clinical trials so far have failed to validate the anxiolytic potential of these drugs, and this line of research has been discontinued (de Haas et al. Citation2009). Alternative methods also include the use of anxiolytics targeting neurotransmitter systems other than the GABAergic one. Indeed, enhancement of serotoninergic activity through buspirone, a 5-HT1A receptor partial agonist, or antidepressants such as selective serotonin uptake inhibitors (SSRIs), comprise a commonly employed method to reduce anxiety symptoms (Lucki Citation1996; Reinhold et al. Citation2011). However, given the relatively slow onset of action of antidepressants, they are often prescribed in combination with other anxiolytics in the first weeks of treatment. Evidence has also shown a role for the glutamatergic system in anxiety, with antagonism of metabotropic glutamate receptors inducing anxiolysis (Dunayevich et al. Citation2008). The antihistamine hydroxyzine, an H1 receptor inverse agonist as well as a 5-HT2A receptor antagonist, also seems effective in the treatment of anxiety (Lader and Scotto Citation1998). Overall, these molecules still appear to carry a GABAergic component, as serotoninergic fibres act on GABA neurons, and as reducing glutamate activity favours general inhibition.
Currently, perhaps the most promising pharmacological alternative for the alleviation of anxiety conditions lies with the neurosteroids. Although they constitute one of the most potent groups of GABAAR modulators, their anxiolytic properties have long been underrated, and they may represent a promising option for treatment with limited side effects. It has been pointed out that many anxiolytics currently prescribed have the ability to promote neurosteroid synthesis, which may mediate part of their efficacy. For instance, diazepam and other benzodiazepines can bind to the TSPO (hence its former appellation ‘peripheral benzodiazepine receptor’), and SSRIs appear to correct the neurosteroid imbalance seen in patients with anxiety (Uzunov et al. Citation1996; Pinna et al. Citation2006).
In addition to being a direct modulator of GABAAR function, EFX is also a TSPO ligand, and, as such, the pleiotropic effects of 3α-reduced neurosteroids are intimately involved in its benefits. In several papers comparing the efficacy of EFX and benzodiazepines, EFX showed interesting properties with limited side effects, which opens the door for a new generation of anxiolytics targeting the TSPO (Nguyen et al. Citation2006; Stein Citation2015). Mitochondrial TSPO ligands may act in almost every cell type of the organism, and the stimulation of neurosteroidogenesis in nervous and peripheral tissues is far from being trivial. For example, rare severe liver injuries and toxidermia have been suspected to be possibly associated with EFX treatment in two recent studies published by French groups (Moch et al. Citation2012; Cottin et al. Citation2016). Although the double-targeting properties of drugs such as EFX show considerable potential, further investigation on the molecular mechanisms underlying TSPO ligand efficacy is still needed. Their better understanding could, inter alia, facilitate the development of synthetic neurosteroid analogues.
7. Conclusion
Despite the variety of agents approved for the relief of anxiety conditions, the search for the optimal drug is still ongoing. Although it is unlikely that one unique compound would have beneficial effects on the whole spectrum of anxiety and adjustment disorders, the recognition of neurosteroids as potent anxiolytics gave rise to novel therapeutic strategies. It is, for example, interesting to note that the production of pregnenolone after TSPO stimulation has been recently suggested to exert a potent inhibition of cannabinoid receptor type 1 (CB1), thus limiting psychotic-like states for this drug of abuse (Busquets-Garcia et al. Citation2017). Following the innovative trail of etifoxine, TSPO ligands are emerging as a promising method for well-tolerated anxiety relief.
Statement of interest
Pierrick Poisbeau received financial support from Biocodex to investigate the molecular mechanisms of action of etifoxine.
Acknowledgements
None.
Additional information
Funding
References
- Akk G, Shu HJ, Wang C, Steinbach JH, Zorumski CF, Covey DF, Mennerick S. 2005. Neurosteroid access to the GABAA receptor. J Neurosci. 25:11605–11613.
- Ali NJ, Olsen RW. 2001. Chronic benzodiazepine treatment of cells expressing recombinant GABA(A) receptors uncouples allosteric binding: studies on possible mechanisms. J Neurochem. 79:1100–1108.
- Aouad M, Charlet A, Rodeau JL, Poisbeau P. 2009. Reduction and prevention of vincristine-induced neuropathic pain symptoms by the non-benzodiazepine anxiolytic etifoxine are mediated by 3alpha-reduced neurosteroids. Pain. 147:54–59.
- Aouad M, Petit-Demouliere N, Goumon Y, Poisbeau P. 2014. Etifoxine stimulates allopregnanolone synthesis in the spinal cord to produce analgesia in experimental mononeuropathy. Eur J Pain. 18:258–268.
- Aouad M, Zell V, Juif PE, Lacaud A, Goumon Y, Darbon P, Lelievre V, Poisbeau P. 2014. Etifoxine analgesia in experimental monoarthritis: a combined action that protects spinal inhibition and limits central inflammatory processes. Pain. 155:403–412.
- Bormann J. 1991. Electrophysiological characterization of diazepam binding inhibitor (DBI) on GABAA receptors. Neuropharmacology. 30:1387–1389.
- Busquets-Garcia A, Soria-Gomez E, Redon B, Mackenbach Y, Vallee M, Chaouloff F, Varilh M, Ferreira G, Piazza PV, Marsicano G. 2017. Pregnenolone blocks cannabinoid-induced acute psychotic-like states in mice. Mol Psychiatry. 22:1594–1603.
- Calhoon GG, Tye KM. 2015. Resolving the neural circuits of anxiety. Nat Neurosci. 18:1394–1404.
- Callachan H, Cottrell GA, Hather NY, Lambert JJ, Nooney JM, Peters JA. 1987. Modulation of the GABAA receptor by progesterone metabolites. Proc R Soc Lond B Biol Sci. 231:359–369.
- Carver CM, Reddy DS. 2016. Neurosteroid structure-activity relationships for functional activation of extrasynaptic deltaGABA(A) receptors. J Pharmacol Exp Ther. 357:188–204.
- Cheng VY, Martin LJ, Elliott EM, Kim JH, Mount HT, Taverna FA, Roder JC, Macdonald JF, Bhambri A, Collinson N, et al. 2006. Alpha5GABAA receptors mediate the amnestic but not sedative-hypnotic effects of the general anesthetic etomidate. J Neurosci. 26:3713–3720.
- Chiara DC, Jayakar SS, Zhou X, Zhang X, Savechenkov PY, Bruzik KS, Miller KW, Cohen JB. 2013. Specificity of intersubunit general anesthetic-binding sites in the transmembrane domain of the human alpha1beta3gamma2 gamma-aminobutyric acid type A (GABAA) receptor. J Biol Chem. 288:19343–19357.
- Chisari M, Shu HJ, Taylor A, Steinbach JH, Zorumski CF, Mennerick S. 2010. Structurally diverse amphiphiles exhibit biphasic modulation of GABAA receptors: similarities and differences with neurosteroid actions. Br J Pharmacol. 160:130–141.
- Choi YM, Kim KH. 2015. Etifoxine for pain patients with anxiety. Korean J Pain. 28:4–10.
- Christian CA, Herbert AG, Holt RL, Peng K, Sherwood KD, Pangratz-Fuehrer S, Rudolph U, Huguenard JR. 2013. Endogenous positive allosteric modulation of GABA(A) receptors by diazepam binding inhibitor. Neuron. 78:1063–1074.
- Collinson N, Kuenzi FM, Jarolimek W, Maubach KA, Cothliff R, Sur C, Smith A, Otu FM, Howell O, Atack JR, et al. 2002. Enhanced learning and memory and altered GABAergic synaptic transmission in mice lacking the alpha 5 subunit of the GABAA receptor. J Neurosci. 22:5572–5580.
- Costa E, Guidotti A. 1991. Diazepam binding inhibitor (DBI): a peptide with multiple biological actions. Life Sci. 49:325–344.
- Cottin J, Gouraud A, Jean-Pastor MJ, Dautriche AD, Boulay C, Geniaux H, Auffret M, Bernard N, Descotes J, Vial T. 2016. Safety profile of etifoxine: a French pharmacovigilance survey. Fundam Clin Pharmacol. 30:147–152.
- Coull JA, Boudreau D, Bachand K, Prescott SA, Nault F, Sik A, De Koninck P, De Koninck Y. 2003. Trans-synaptic shift in anion gradient in spinal lamina I neurons as a mechanism of neuropathic pain. Nature. 424:938–942.
- Crestani F, Keist R, Fritschy JM, Benke D, Vogt K, Prut L, Bluthmann H, Mohler H, Rudolph U. 2002. Trace fear conditioning involves hippocampal alpha5 GABA(A) receptors. Proc Natl Acad Sci U S A. 99:8980–8985.
- Crestani F, Low K, Keist R, Mandelli M, Mohler H, Rudolph U. 2001. Molecular targets for the myorelaxant action of diazepam. Mol Pharmacol. 59:442–445.
- de Haas SL, Franson KL, Schmitt JA, Cohen AF, Fau JB, Dubruc C, van Gerven JM. 2009. The pharmacokinetic and pharmacodynamic effects of SL65.1498, a GABA-A alpha2,3 selective agonist, in comparison with lorazepam in healthy volunteers. J Psychopharmacol. 23:625–632.
- Dias R, Sheppard WF, Fradley RL, Garrett EM, Stanley JL, Tye SJ, Goodacre S, Lincoln RJ, Cook SM, Conley R, et al. 2005. Evidence for a significant role of alpha 3-containing GABAA receptors in mediating the anxiolytic effects of benzodiazepines. J Neurosci. 25:10682–10688.
- do Rego JL, Vaudry D, Vaudry H. 2015. The non-benzodiazepine anxiolytic drug etifoxine causes a rapid, receptor-independent stimulation of neurosteroid biosynthesis. PLoS One. 10:e0120473.
- Dunayevich E, Erickson J, Levine L, Landbloom R, Schoepp DD, Tollefson GD. 2008. Efficacy and tolerability of an mGlu2/3 agonist in the treatment of generalized anxiety disorder. Neuropsychopharmacology. 33:1603–1610.
- Fan J, Liu J, Culty M, Papadopoulos V. 2010. Acyl-coenzyme A binding domain containing 3 (ACBD3; PAP7; GCP60): an emerging signaling molecule. Prog Lipid Res. 49:218–234.
- Ffrench-Mullen JM, Barker JL, Rogawski MA. 1993. Calcium current block by (-)-pentobarbital, phenobarbital, and CHEB but not (+)-pentobarbital in acutely isolated hippocampal CA1 neurons: comparison with effects on GABA-activated Cl- current. J Neurosci. 13:3211–3221.
- Frye CA, Walf AA. 2004. Estrogen and/or progesterone administered systemically or to the amygdala can have anxiety-, fear-, and pain-reducing effects in ovariectomized rats. Behav Neurosci. 118:306–313.
- Girard C, Liu S, Cadepond F, Adams D, Lacroix C, Verleye M, Gillardin JM, Baulieu EE, Schumacher M, Schweizer-Groyer G. 2008. Etifoxine improves peripheral nerve regeneration and functional recovery. Proc Natl Acad Sci USA. 105:20505–20510.
- Girard P, Pansart Y, Gillardin JM. 2009. Preventive and curative effects of etifoxine in a rat model of brain oedema. Clin Exp Pharmacol Physiol. 36:655–661.
- Griebel G, Perrault G, Simiand J, Cohen C, Granger P, Depoortere H, Francon D, Avenet P, Schoemaker H, Evanno Y, et al. 2003. SL651498, a GABAA receptor agonist with subtype-selective efficacy, as a potential treatment for generalized anxiety disorder and muscle spasms. CNS Drug Rev. 9:3–20.
- Guidotti A, Forchetti CM, Corda MG, Konkel D, Bennett CD, Costa E. 1983. Isolation, characterization, and purification to homogeneity of an endogenous polypeptide with agonistic action on benzodiazepine receptors. Proc Natl Acad Sci USA. 80:3531–3535.
- Gutierrez-Garcia AG, Contreras CM. 2013. Chapter 2, Anxiety: an adaptive emotion. In: Durbano F, editor. Mental and behavioral disorders and diseases of the nervous system: new insight into anxiety disorders. Rijeka: InTech; p. 21–37.
- Hamon A, Morel A, Hue B, Verleye M, Gillardin JM. 2003. The modulatory effects of the anxiolytic etifoxine on GABA(A) receptors are mediated by the beta subunit. Neuropharmacology. 45:293–303.
- Heikkinen AE, Moykkynen TP, Korpi ER. 2009. Long-lasting modulation of glutamatergic transmission in VTA dopamine neurons after a single dose of benzodiazepine agonists. Neuropsychopharmacology. 34:290–298.
- Hosie AM, Wilkins ME, da Silva HM, Smart TG. 2006. Endogenous neurosteroids regulate GABAA receptors through two discrete transmembrane sites. Nature. 444:486–489.
- Johns MW. 1977. Self-poisoning with barbiturates in England and Wales during 1959-74. Br Med J. 1:1128–1130.
- Juif PE, Melchior M, Poisbeau P. 2015. Characterization of the fast GABAergic inhibitory action of etifoxine during spinal nociceptive processing in male rats. Neuropharmacology. 91:117–122.
- Kruse HJ, Kuch H. 1985. Etifoxine: evaluation of its anticonvulsant profile in mice in comparison with sodium valproate, phenytoin and clobazam. Arzneimittelforschung. 35:133–135.
- Kruse HJ, Kuch H. 1986. Potentiation of clobazam's anticonvulsant activity by etifoxine, a non-benzodiazepine tranquilizer, in mice. Comparison studies with sodium valproate. Arzneimittelforschung. 36:1320–1322.
- Lader M, Scotto JC. 1998. A multicentre double-blind comparison of hydroxyzine, buspirone and placebo in patients with generalized anxiety disorder. Psychopharmacology (Berl). 139:402–406.
- Leroy C, Poisbeau P, Keller AF, Nehlig A. 2004. Pharmacological plasticity of GABA(A) receptors at dentate gyrus synapses in a rat model of temporal lobe epilepsy. J Physiol. 557:473–487.
- Longone P, di Michele F, D'Agati E, Romeo E, Pasini A, Rupprecht R. 2011. Neurosteroids as neuromodulators in the treatment of anxiety disorders. Front Endocrin. 2:55.
- Loomis WF, Behrens MM, Williams ME, Anjard C. 2010. Pregnenolone sulfate and cortisol induce secretion of acyl-CoA-binding protein and its conversion into endozepines from astrocytes. J Biol Chem. 285:21359–21365.
- Low K, Crestani F, Keist R, Benke D, Brunig I, Benson JA, Fritschy JM, Rulicke T, Bluethmann H, Mohler H, et al. 2000. Molecular and neuronal substrate for the selective attenuation of anxiety. Science. 290:131–134.
- Lucki I. 1996. Serotonin receptor specificity in anxiety disorders. J Clin Psychiatry. 57(Suppl 6):5–10.
- McKernan RM, Rosahl TW, Reynolds DS, Sur C, Wafford KA, Atack JR, Farrar S, Myers J, Cook G, Ferris P, et al. 2000. Sedative but not anxiolytic properties of benzodiazepines are mediated by the GABA(A) receptor alpha1 subtype. Nat Neurosci.Neurosci3:587–592.
- Moch C, Rocher F, Laine P, Lacotte J, Biour M, Gouraud A, Bernard N, Descotes J, Vial T. 2012. Etifoxine-induced acute hepatitis: a case series. Clin Res Hepatol Gastroenterol. 36:e85–e88.
- Mohler H, Fritschy JM, Rudolph U. 2002. A new benzodiazepine pharmacology. J Pharmacol Exp Ther. 300:2–8.
- Nguyen N, Fakra E, Pradel V, Jouve E, Alquier C, Le Guern ME, Micallef J, Blin O. 2006. Efficacy of etifoxine compared to lorazepam monotherapy in the treatment of patients with adjustment disorders with anxiety: a double-blind controlled study in general practice. Hum Psychopharmacol Clin Exp. 21:139–149.
- Nothdurfter C, Rammes G, Baghai TC, Schule C, Schumacher M, Papadopoulos V, Rupprecht R. 2012. Translocator protein (18 kDa) as a target for novel anxiolytics with a favourable side-effect profile. J Neuroendocrinol. 24:82–92.
- Olsen RW. 2018. GABAA receptor: Positive and negative allosteric modulators. Neuropharmacology. https://doi.org/10.1016/j.neuropharm.2018.01.036
- Olsen RW, Sieghart W. 2009. GABA A receptors: subtypes provide diversity of function and pharmacology. Neuropharmacology. 56:141–148.
- Patchev VK, Shoaib M, Holsboer F, Almeida OF. 1994. The neurosteroid tetrahydroprogesterone counteracts corticotropin-releasing hormone-induced anxiety and alters the release and gene expression of corticotropin-releasing hormone in the rat hypothalamus. Neuroscience. 62:265–271.
- Pinna G, Costa E, Guidotti A. 2006. Fluoxetine and norfluoxetine stereospecifically and selectively increase brain neurosteroid content at doses that are inactive on 5-HT reuptake. Psychopharmacology (Berl). 186:362–372.
- Poisbeau P, Feltz P, Schlichter R. 1997. Modulation of GABAA receptor-mediated IPSCs by neuroactive steroids in a rat hypothalamo-hypophyseal coculture model. J Physiol. 500: 475–485.
- Poisbeau P, Keller AF, Aouad M, Kamoun N, Groyer G, Schumacher M. 2014. Analgesic strategies aimed at stimulating the endogenous production of allopregnanolone. Front Cell Neurosci. 8:174.
- Poisbeau P, Williams SR, Mody I. 1997. Silent GABAA synapses during flurazepam withdrawal are region-specific in the hippocampal formation. J Neurosci. 17:3467–3475.
- Ravikumar B, Crawford D, Dellovade T, Savinainen A, Graham D, Liere P, Oudinet JP, Webb M, Hering H. 2016. Differential efficacy of the TSPO ligands etifoxine and XBD-173 in two rodent models of Multiple Sclerosis. Neuropharmacology. 108:229–237.
- Reinhold JA, Mandos LA, Rickels K, Lohoff FW. 2011. Pharmacological treatment of generalized anxiety disorder. Expert Opin Pharmacother. 12:2457–2467.
- Rudolph U, Crestani F, Benke D, Brunig I, Benson JA, Fritschy JM, Martin JR, Bluethmann H, Mohler H. 1999. Benzodiazepine actions mediated by specific gamma-aminobutyric acid(A) receptor subtypes. Nature. 401:796–800.
- Rupprecht R, Papadopoulos V, Rammes G, Baghai TC, Fan J, Akula N, Groyer G, Adams D, Schumacher M. 2010. Translocator protein (18 kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat Rev Drug Discov. 9:971–988.
- Sammler E, Titz S, Hormuzdi S. 2015. Neuronal chloride transport tuning. Lancet. 385(Suppl 1):S85.
- Schlichter R, Rybalchenko V, Poisbeau P, Verleye M, Gillardin J. 2000. Modulation of GABAergic synaptic transmission by the non-benzodiazepine anxiolytic etifoxine. Neuropharmacology. 39:1523–1535.
- Schule C, Eser D, Baghai TC, Nothdurfter C, Kessler JS, Rupprecht R. 2011. Neuroactive steroids in affective disorders: target for novel antidepressant or anxiolytic drugs?. Neuroscience. 191:55–77.
- Servant D, Graziani PL, Moyse D, Parquet PJ. 1998. [Treatment of adjustment disorder with anxiety: efficacy and tolerance of etifoxine in a double-blind controlled study]. Encephale. 24:569–574.
- Shu HJ, Eisenman LN, Jinadasa D, Covey DF, Zorumski CF, Mennerick S. 2004. Slow actions of neuroactive steroids at GABAA receptors. J Neurosci Neurosci. 24:6667–6675.
- Simon-O'Brien E, Gauthier D, Riban V, Verleye M. 2016. Etifoxine improves sensorimotor deficits and reduces glial activation, neuronal degeneration, and neuroinflammation in a rat model of traumatic brain injury. J Neuroinflammation. 13:203.
- Stein DJ. 2015. Etifoxine versus alprazolam for the treatment of adjustment disorder with anxiety: a randomized controlled trial. Adv Ther. 32:57–68.
- Stell BM, Brickley SG, Tang CY, Farrant M, Mody I. 2003. Neuroactive steroids reduce neuronal excitability by selectively enhancing tonic inhibition mediated by delta subunit-containing GABAA receptors. Proc Natl Acad Sci U S A. 100:14439–14444.
- Tan KR, Brown M, Labouebe G, Yvon C, Creton C, Fritschy JM, Rudolph U, Luscher C. 2010. Neural bases for addictive properties of benzodiazepines. Nature. 463:769–774.
- Tan KR, Rudolph U, Luscher C. 2011. Hooked on benzodiazepines: GABAA receptor subtypes and addiction. Trends Neurosci. 34:188–197.
- Twyman RE, Macdonald RL. 1992. Neurosteroid regulation of GABAA receptor single-channel kinetic properties of mouse spinal cord neurons in culture. J Physiol. 456:215–245.
- Ugale RR, Sharma AN, Kokare DM, Hirani K, Subhedar NK, Chopde CT. 2007. Neurosteroid allopregnanolone mediates anxiolytic effect of etifoxine in rats. Brain Res. 1184:193–201.
- Uzunov DP, Cooper TB, Costa E, Guidotti A. 1996. Fluoxetine-elicited changes in brain neurosteroid content measured by negative ion mass fragmentography. Proc Natl Acad Sci U S A. 93:12599–12604.
- Verleye M, Akwa Y, Liere P, Ladurelle N, Pianos A, Eychenne B, Schumacher M, Gillardin JM. 2005. The anxiolytic etifoxine activates the peripheral benzodiazepine receptor and increases the neurosteroid levels in rat brain. Pharmacol Biochem Behav.82:712–720.
- Verleye M, Dumas S, Heulard I, Krafft N, Gillardin JM. 2011. Differential effects of etifoxine on anxiety-like behaviour and convulsions in BALB/cByJ and C57BL/6J mice: any relation to overexpression of central GABAA receptor beta2 subunits? Eur Neuropsychopharmacol. 21:457–470.
- Verleye M, Gillardin JM. 2004. Effects of etifoxine on stress-induced hyperthermia, freezing behavior and colonic motor activation in rats. Physiol Behav. 82:891–897.
- Verleye M, Heulard I, Gillardin JM. 2009. The anxiolytic etifoxine protects against convulsant and anxiogenic aspects of the alcohol withdrawal syndrome in mice. Alcohol. 43:197–206.
- Verleye M, Schlichter R, Gillardin JM. 1999. Interactions of etifoxine with the chloride channel coupled to the GABA(A) receptor complex. Neuroreport10:3207–3210.
- Wang M, He Y, Eisenman LN, Fields C, Zeng CM, Mathews J, Benz A, Fu T, Zorumski E, Steinbach JH, et al. 2002. 3beta -hydroxypregnane steroids are pregnenolone sulfate-like GABA(A) receptor antagonists. J Neurosci. 22:3366–3375.
- Wolf L, Bauer A, Melchner D, Hallof-Buestrich H, Stoertebecker P, Haen E, Kreutz M, Sarubin N, Milenkovic VM, Wetzel CH, et al. 2015 . Enhancing neurosteroid synthesis-relationship to the pharmacology of translocator protein (18 kDa) (TSPO) ligands and benzodiazepines. Pharmacopsychiatry. 48:72–77.
- Zeitler A, Kamoun N, Goyon S, Wahis J, Charlet A, Poisbeau P, Darbon P. 2016. Favouring inhibitory synaptic drive mediated by GABA(A) receptors in the basolateral nucleus of the amygdala efficiently reduces pain symptoms in neuropathic mice. Eur J Neurosci. 43:1082–1088.
- Zhou X, He X, He B, Zhu Z, Zheng C, Xu J, Jiang L, Gu L, Zhu J, Zhu Q, et al. 2013. Etifoxine promotes glialderived neurotrophic factorinduced neurite outgrowth in PC12 cells. Mol Med Rep. 8:75–80.
- Zhou X, He B, Zhu Z, He X, Zheng C, Xu J, Jiang L, Gu L, Zhu J, Zhu Q, et al. 2014. Etifoxine provides benefits in nerve repair with acellular nerve grafts. Muscle Nerve. 50:235–243.