Abstract
A series new thermally stable and organosoluble polyamides (PAs) 5a–f and copolyamides 10a–f with good inherent viscosities were synthesized from the direct polycondensation reaction of 1,2-bis(4-carboxyphenoxy)ethane 3 with aromatic diamines 4a–f and from the diacid 3 as a first diacid monomer and 4,4′-bis(1,3-diphenoxypropane) diacrylic acid 9 as a second diacid monomer with aromatic diamines 4a–f, respectively. The dicarboxylic acid 9 was synthesized from the two step reactions. At first 4,4′-bis(1,3-diphenoxypropane) dialdehyde 8 was synthesized from 1,3-dibromopropane 6 and 4-hydroxybenzaldehyde 7, then 4,4′-bis(1,2-diphenoxyethane) diacrylic acid 9 was synthesized from dialdehyde 8 and malonic acid in a solvent free reaction. All of the above polymers were fully characterized by 1H NMR, Fourier transform infrared, elemental analysis, inherent viscosity, solubility tests, differential scanning calorimeter, thermogravimetric analysis, and derivative of thermogravimetric. The resulted PAs have shown admirable good inherent viscosities and thermal stability and solubility.
Introduction
Aromatic polyamides have excellent mechanical properties and thermal stability which make them useful as high-performance materials for advanced technologies and usually they are synthesized of aromatic diamines and aromatic dicarboxylic acids Citation[1,2]. However, applications of polyamides (PAs) are often restricted by problems in their fabrication such as poor solubility and high softening or melting temperatures caused by high crystallinity and high stiffness of the polymer backbone lead to difficult processability of resulting PAs.
Many efforts have been made to create structurally modified aromatic polyamides having better solubility and processability. The introduction of flexible chains into the polyamide backbone Citation[3], synthesis of PAs with noncoplaner unit in the polymer chains Citation[4], preparation of copolymers such as poly(amide-imide)s Citation[5–8], poly(ester-imide)s Citation[9], and poly(amide-ester-imide)s, Citation[10] and the introduction of bulky side groups into the polymer chains Citation[11–13] resulted in a series of modified PAs.
Aromatic polymers that contain aryl ether linkages generally have lower glass transition temperatures, greater chain flexibility, and tractability compared to their corresponding polymers without these groups in the chain Citation[14–16]. The lower glass transition temperatures and also improved solubility are attributed to the flexible linkages that provide a polymer chain with a lower energy of internal rotation Citation[17].
A triphenyl phosphite (TPP)-activated polycondensation (phosphorylation reaction) technique for the synthesis of PAs was reported by Yamazaki et al. Citation[18]. We describe here the preparation and basic characterization of thermally stable and organosoluble of a series PAs 5a–f copolyamides (CoPAs) 10a–f based on ether linkages as a flexible group from the reaction of 1,2-bis(4-carboxyphenoxy)ethane 3 with aromatic diamines 4a–f and from the diacid 3 as a first diacid monomer and 4,4′-bis(1,3-diphenoxypropane) diacrylic acid 9 as a second diacid monomer with aromatic diamines 4a–f, respectively, in a medium consisting of N-methyl-2-pyrrolidone (NMP)/TPP/calcium chloride (CaCl2)/pyridine (Py) system.
Experimental
Materials
1,2-dibromo ethane 1, 1,3-dibromopropane 6, 4-hydroxybenzaldehyde 7, 4-hydroxybenzoic acid 2, 4,4′-diaminodiphenylether 4a, 4,4′-diaminodiphenylsullfone 4b, 3,3′-diaminodiphenylsulfone 4c, 1,4-phenylenediamine 4d, 1,5-naphthalenediamine 4e, 4,4′-diaminobenzidin 4f, malonic acid (Merck), and tosyl chloride (TsCl; Merck) were used without previous purification. Solvent: N-methyl-2-pyrrolidone (NMP; Fluka), pyridine (Acros), triphenyl phosphite (TPP; Merck), and N,N-dimethylformamide (DMF; Merck) were used as received. Commercially available calcium chloride (CaCl2; Merck) was dried under vacuum at 150 °C for 6 h.
Techniques
1H and 13C NMR spectra were recorded on a Bruker 300 MHz instrument (Germany) and with a Bruker Avance III 500 spectrometer (Rheinstetten, Germany) operating at 500 MHz (1H). The DMSO-d 6 was used as the solvent and the solvent signal was used for internal calibration (DMSO-d 6: δ(1H) = 2.5 ppm). Fourier transform infrared (FTIR) spectra were recorded on Galaxy series FTIR 5000 spectrophotometer (England). Spectra of solid were performed by using KBr pellets. Vibration transition frequencies were reported in wave number (cm−1). Band intensities were assigned as weak (w), medium (m), shoulder (sh), strong (s), and broad (br). Inherent viscosities were measured by a standard procedure by using a Technico Regd Trad Mark Viscometer. Thermal gravimetric analysis (TGA and DTG) data for polymers were taken on a Mettler TA4000 System under N2 atmosphere at a rate of 10 °C min−1 and differential scanning calorimeter (DSC) was conducted with a DSC Metller 110 (Switzerland) at a heating rate of 10 °C min−1 in a nitrogen atmosphere. Elemental analyses were performed by Vario EL equipment.
Monomer synthesis
Synthesis of 1,2-bis(4-carboxyphenoxy)ethane 3
In a 250 mL round-bottomed flask with dropping funnel fitted with a stirring bar were placed (4.60 g, 33.3 mmol) of 4-hydroxy benzoic acid 2 and (2.60 g, 65.0 mmol) sodium hydroxide in 14.0 mL H2O. Then 16.8 mmol 1,2-dibromo ethane 1 was added into the reaction mixture slowly with stirring and the reaction mixture was refluxed for 3.5 h. After that (0.66 g, 16.5 mmol) NaOH was added and refluxing continued for 2 h. Then the heating was removed, the stirring continued at room temperature for overnight. After that the white precipitate was filtered and washed with 10 mL methanol, the solid was dissolved in 34 mL H2O, by adding a solution of H2SO4 (6 N) a white solid was precipitated, washed with the cold water, and filtered at room temperature until 3.36 g (67%) of white product was obtained. FTIR (KBr): 2500–3400 (s, br), 1680 (s, br), 1606 (s), 1514 (s), 1435 (s), 1303 (m), 1251 (m), 1167 (m), 1047 (s), 945 (m), 844 (s), 769 (m), 646 (m), 553 (m) cm−1. 1H NMR (DMSO-d 6, TMS) δ: 4.41 (s, 4H), 7.06–7.08 (d, 4H), 7.88–7.91 (d, 4H), 12.64 (s, br, 2H) ppm. 13C NMR (300 MHz, DMSO-d 6) δ: 167.4, 162.2, 131.8, 123.3, 114.7, 66.8 ppm. Elemental analysis: calculated for C16H14O6: C, 63.57; H, 4.67; found: C, 62.29; H, 4.62.
Synthesis of 4,4′-bis(1,3-diphenoxypropane) dialdehyde 8
In a 250 mL round-bottomed flask with dropping funnel fitted with a stirring bar were placed (4.06 g, 33.3 mmol) of 4-hydroxybenzaldehyde 2 and (2.60 g, 65.0 mmol) sodium hydroxide in 14.0 mL H2O. Then 16.8 mmol 1,3-dibromopropane 6 was added into the reaction mixture slowly with stirring and the reaction mixture was refluxed for 3.5 h. After that (0.66 g, 16.5 mmol) NaOH was added and refluxing continued for 2 h. Then the heating was removed, the stirring continued at room temperature for overnight. After that the white precipitate was filtered and washed with 15 mL methanol, the solid was dissolved in 34 mL H2O, by adding a solution of H2SO4 (6 N) a white solid was precipitated, washed with the cold water, and filtered at room temperature until 6.23 g (66%) of white product was obtained. Mp: 202–204 °C, FTIR (KBr): 3025 (w), 2927 (m), 2919 (w), 1812 (s), 1700 (s), 1682 (s), 1671 (s), 1595 (m), 1410 (s), 1381 (s), 1290 (s), 1169 (m), 1035 (w), 844 (m), 770 (m) cm−1. 1H NMR (DMSO-d 6, TMS) δ: 9.86 (s, 2H), 7.48–7.87 (d, 4H), 7.14–7.16 (d, 4H), 4.27 (t, 4H), 2.24 (m, 2H) ppm.
Synthesis of 4,4′-bis(1,3-diphenoxypropane) diacrylic acid 9
In a 100 mL beaker were placed (1.57 g, 5.55 mmol) of 4,4′-bis(1,2-diphenoxypropane) dialdehyde 8 and (1.15 g, 11.1 mmol) malonic acid in 1 mL morpholine. The mixture compounds were heated until completely melted. Then the heating was removed and 20 mL HCl 5% was added in the reaction mixture slowly with stirring and the stirring continued at room temperature for 2 h. After that the white precipitate was filtered and washed with 20 mL H2O and washed with the cold water and filtered at room temperature until 1.88 g (92%) white product was obtained. Mp: 325–327 °C, FTIR (KBr): 2400–3500 (s, br), 1690 (s), 1628 (s), 1601 (s), 1510 (s), 1427 (s), 1311 (s, sh), 1217 (m), 1172 (m), 1018 (s, br), 827 (m), 682 (w), 509 (w) cm−1. 1H NMR (DMSO-d 6, TMS) δ: 12.2 (s, br, 2H), 7.50–7.61 (m, 6H), 5.99 (d, 4H), 6.34 (d, 2H), 4.16 (t, 4H), 2.17 (m, 2H) ppm. 13C NMR (DMSO-d 6) δ: 168.3, 160.6, 144.1, 130.4, 127.3, 116.9, 115.2, 64.8, 28.9 ppm. Elemental analysis: calculated for C21H20O6: C, 68.47; H, 5.47, found: C, 68.41; H, 5.46.
Polymer synthesis
Synthesis of polyamides 5a–f
The PAs 5a–f were synthesized by direct polycondensation reaction, as an example the preparation of PA 5a explained in the following. The PA 5a was prepared from the reaction of 1,2-bis(4-carboxyphenoxy)ethane 3 with 4,4′-diaminodiphenylether 4a.
The 0.2 g (0.662 mmol) dicarboxylic acid 3, 0.122 g (0.662 mmol) 4,4′-diaminodiphenylether 4a, 0.1 g (0.9 mmol), CaCl2 0.84 mL, (3.00 mmol) TPP, 0.1 mL of pyridine, and 2 mL NMP were placed into a 25-mL round-bottomed flask, which was fitted with a stirring bar. The reaction mixture was heated under reflux on an oil bath at 120 °C for 8 h. Then, the reaction mixture was poured into 50 mL of methanol and the precipitated polymer was collected by filtration and washed thoroughly with hot methanol and dried at 60°C for 12 h under vacuum to leave 0.281 g (91%) of white solid polymer 5a.
Polymer 5a
FTIR (KBr): FTIR (KBr): 3277 (w), 3015 (m), 2925 (m), 1628(s), 1549 (m), 1478 (m), 1435 (m), 1388 (m), 1265 (m), 1165 (m), 1060 (m), 952 (w), 729 (m), 652 (m) cm−1. Elemental analysis: calculated for C28H22N2O5: C, 72.09; H, 4.75; N, 6.01; found: C, 71.54; H, 4.55; N, 589.
Polymer 5b
FTIR (KBr): 3244 (w), 3085 (m), 2988 (m), 1685 (s), 1620 (s), 1578 (m), 1539 (m), 1480 (m), 1420 (m), 1380 (m), 1277 (m), 1170 (m), 1079 (m), 950 (w), 729 (m), 699 (m) cm−1. Elemental analysis: calculated for C28H22N2O6S: C, 65.36; H, 4.31; N, 5.44; found: C, 64.22; H, 4.22; N, 5.31.
Polymer 5c
FTIR (KBr): 3311 (w), 3022 (m), 2870 (m), 1680 (s), 1613(s), 1522 (m), 1540 (m), 1484 (m), 1427 (m), 1383 (m), 1279 (m), 1179 (m), 1071 (m), 962 (w), 727 (m), 650 (m) cm−1. Elemental analysis: calculated for C28H22N2O6S: C, 65.36; H, 4.31; N, 5.44; found: C, 64.11; H, 4.28; N, 5.42.
Polymer 5d
FTIR (KBr): 3277 (w), 3015 (m), 2925 (m), 1628(s), 1549 (m), 1478 (m), 1435 (m), 1388 (m), 1265 (m), 1165 (m), 1060 (m), 952 (w), 729 (m), 652 (m) cm−1. Elemental analysis: calculated for C22H18N2O4: C, 70.58; H, 4.85; N, 7.48; found: C, 68.89; H, 4.76; N, 7.45.
Polymer 5e
FTIR (KBr): 3277 (w), 3015 (m), 2925 (m), 1628(s), 1549 (m), 1478 (m), 1435 (m), 1388 (m), 1265 (m), 1165 (m), 1060 (m), 952 (w), 729 (m), 652 (m) cm−1. Elemental analysis: calculated for C26H20N2O4: C, 73.57; H, 4.75; N, 6.60; found: C, 72.11; H, 4.31; N, 6.24.
Polymer 5f
FTIR (KBr): 3294 (m), 3059 (w), 2962 (m), 1682 (s), 1589 (m), 1541 (m), 1479 (m), 1415 (m), 1375 (m), 1303 (m), 1246 (m), 1167 (m), 1082 (m), 939 (w), 725 (m), 690 (m) cm−1. Elemental analysis: calculated for C28H22N2O4: C, 74.65; H, 4.92; N, 6.22; found: C, 73.08; H, 4.53; N, 6.13.
Synthesis of CoPolyamides 10a–f
In a 100 mL round-bottomed flask were placed a mixture of 1,2-bis(4-carboxyphenoxy)ethane 3 (0.326 mmol), second diacid 9 (0.326 mmol), diamines 4a–f (0.652 mmol), 0.10 g of CaCl2, 0.84 mL of TPP, 0.2 mL of pyridine, and 1.8 mL of NMP. The mixture was heated for 1 h at 60 °C and 2 h at 90 °C and then refluxed at 130 °C for 7 h until a viscose solution was formed. Then it was cooled to room temperature and 30 mL of methanol was added to the reaction mixture. The precipitate was formed, filtered off, and washed with methanol. The resulting polymers 10a–f were dried under vacuum.
Polymer 10a
FTIR (KBr): 3340 (m), 2957 (m), 1709 (s), 1589 (m), 1541 (m), 1481 (m), 1384 (m), 1186 (m), 690 (w), 501 (w) cm−1. Elemental analysis: calculated for C61H50N4O10: C, 73.33; H, 5.04; N, 5.61; found: C, 71.86; H, 4.98; N, 5.57.
Polymer 10b
FTIR (KBr): 3320 (w), 3075 (m), 2987 (m), 1685 (s), 1622 (s), 1578 (m), 1539 (m), 1480 (m), 1424 (m), 1380 (m), 1278 (m), 1170 (m), 1079 (m), 950 (w), 729 (m), 699 (m) cm−1. Elemental analysis: calculated for C61H50N4O12S2: C, 66.90; H, 4.60; N, 5.12; found: C, 65.25; H, 4.51; N, 5.05.
Polymer 10c
FTIR (KBr): 3288 (w), 3022 (m), 2970 (m), 1688 (s), 1615(s), 1522 (m), 1540 (m), 1483 (m), 1427 (m), 1380 (m), 1279 (m), 1180 (m), 1071 (m), 962 (w), 727 (m), 650 (m) cm−1. Elemental analysis: calculated for C61H50N4O12S2: C, 66.90; H, 4.60; N, 5.12; found: C, 65.17; H, 4.48; N, 5.02.
Polymer 10d
FTIR (KBr): 3277 (w), 3015 (m), 2925 (m), 1706 (s), 1550 (m), 1481 (m), 1435 (m), 1385 (m), 1264 (m), 1165 (m), 1064 (m), 952 (w), 730 (m), 652 (m) cm−1. Elemental analysis: calculated for C49H42N4O8: C, 72.22; H, 5.20; N, 6.88; found: C, 71.37; H, 5.13; N, 6.81.
Polymer 10e
FTIR (KBr): 3277 (w), 3015 (m), 2925 (m), 1711 (s), 1549 (m), 1479 (m), 1435 (m), 1388 (m), 1268 (m), 1165 (m), 1062 (m), 952 (w), 731 (m), 655 (m) cm−1. Elemental analysis: calculated for C57H48N4O8: C, 78.82; H, 5.07; N, 6.12; found: C, 77.09; H, 5.01; N, 6.12.
Polymer 10f
FTIR (KBr): 3322 (w), 3015 (m), 2985 (m), 1694 (s), 1549 (m), 1479 (m), 1435 (m), 1388 (m), 1259 (m), 1165 (m), 1068 (m), 952 (w), 726 (m), 651 (m) cm−1. Elemental analysis: calculated for C61H50N4O8: C, 75.76; H, 5.21; N, 5.79; found: C, 74.19; H, 5.14; N, 5.73.
Results and discussion
Monomer synthesis
1,2-bis(4-carboxyphenoxy)ethane 3
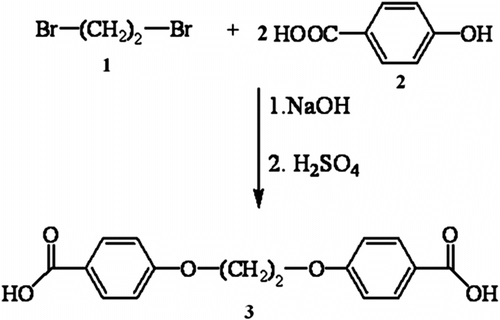
1,2-bis(4-carboxyphenoxy)ethane 3 was prepared according to our previous works Citation[19]. This compound was prepared from the reaction of two equivalents 4-hydroxy benzoic acid 2 and one equivalents 1,2-dibromoethane 1 in a solution of sodium hydroxide (Scheme ).
Scheme 1 Synthesis of diacid 3.
4,4′-Bis(1,3-diphenoxypropane) diacrylic acid 9
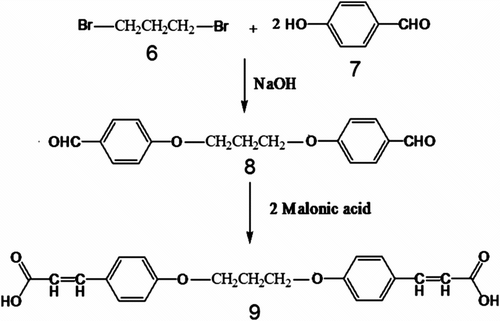
Dicarboxylic acid 5 was synthesized by using two step reactions. At first 4,4′-bis(1,3-diphenoxypropane) dialdehyde 8 was prepared from the reaction of one equimolar 1,3-dibromopropane 6 and two equimolar 4-hydroxybenzaldehyde 7 in aqueous solution of sodium hydroxide. Then dialdehyde compound 8 was reacted with 2 equimolar of malonic acid in a solvent free reaction. Diacid 9 was synthesized by this method with good yield and facile condition (Scheme ).
Scheme 2 Synthesis of dicarboxylic acid 9.
The chemical structure and purity of dialdehyde compound 8 were confirmed with 1H NMR and FTIR spectroscopy and dicarboxylic acid compound 9 was proved with elemental analysis, FTIR, 1H NMR, and 13C NMR spectroscopy. The measured results in elemental analyses of these compounds were closely corresponded to the calculated ones, demonstrating that the expected compounds were obtained.
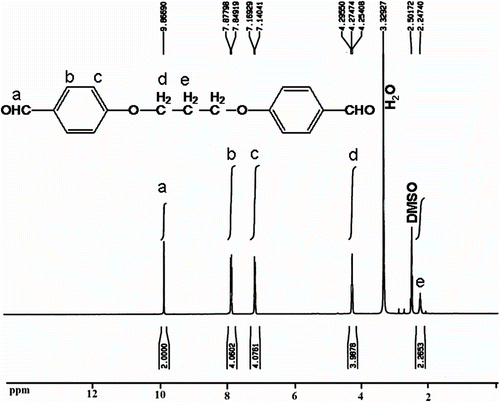
The 1H NMR spectrum of 4,4′-bis(1,3-diphenoxypropane) dialdehyde 8 shows a singlet peak at 9.86 ppm related to aldehyde protons and peaks at 7.84–7.87 and 7.14–7.16 ppm (J = 8.6 Hz) were assigned to H(b) and H(c) related to aromatic protons (Figure ).
Figure 1 1H NMR spectrum of 4,4′-bis(1,3-diphenoxypropane) dialdehyde 8.
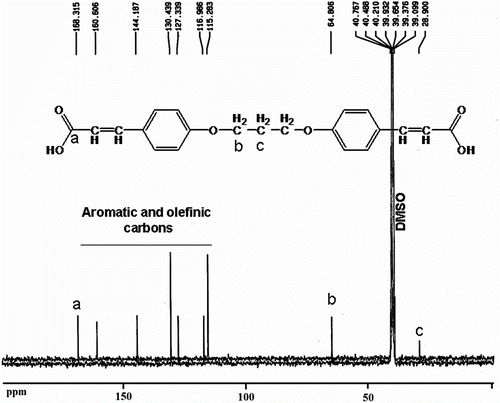
The FTIR spectrum of dicarboxylic acid 9 shows a broad peak at 2400–3500 cm−1, which were assigned to the OH related to carboxylic acid groups in this compound and strong peak at 1690 cm−1 related to carbonyl groups (Figure ). Also 1H NMR spectrum of dicarboxylic acid 9 showed that two peaks as a doublet of doublet at 7.56 ppm and 6.97 were assigned to the H(b) and H(d) related to aromatic protons and two peaks as a doublet of doublet at 7.50 and 6.34 ppm were assigned to the H(c) and H(e) related to olefinic protons. A broad singlet peak at 12.1 ppm was related to carboxylic acid protons (Figure ). Also, the 13C NMR spectrum of dicarboxylic acid 9 showed nine different carbon atoms (Figure ).
Figure 2 FTIR spectrum of 4,4′-bis(1,3-diphenoxypropane) diacrylic acid 9.
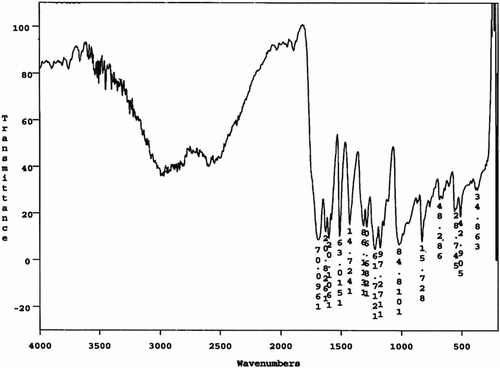
Figure 3 1H NMR spectrum of 4,4′-bis(1,3-diphenoxypropane) diacrylic acid 9.
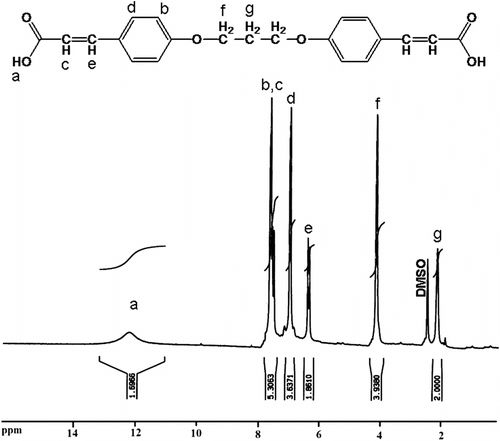
Figure 4 13C NMR spectrum of 4,4′-bis(1,3-diphenoxypropane) diacrylic acid 9.
Polymer synthesis
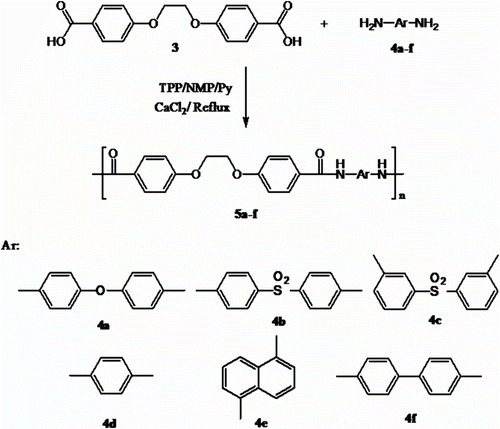
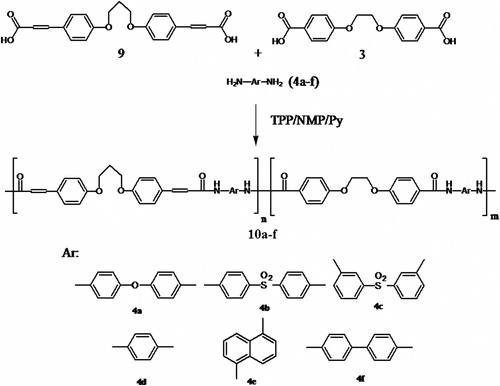
In this article, we synthesized organosoluble and thermally stable PAs 5a–f containing ether linkages in the backbone by the reactions of diacid 3 with six aromatic diamines 4a–f by direct polycondensation (Scheme ) and CoPAs 10a–f containing ether linkages and olefinic groups in the backbone by the reaction of diacid 3 as first diacid monomer and diacid 9 as second diacid monomer and aromatic diamines 4a–f (Scheme ) in a medium consisting of NMP/TPP/CaCl2/ pyridine (py) system.
Scheme 3 Synthesis of PAs 5a–f.
Scheme 4 Synthesis of CoPAs 10a–f.
For synthesis of these polymers, we used from direct polycondensation TPP/Py/CaCl2 as activating agent that is shown in Schemes and . The entire polycondensation reaction readily proceeds in a homogeneous solution, tough and stringy precipitates were formed when the viscous PAs and CoPAs solution was obtained in good yields. Also the resulting polymers have a range of color between white and cream (Table ).
Table 1. Syntheses and some physical properties of PAs 5a–f and CoPAs 10a–f.
Polymer characterization
The structure of these polymers was confirmed as PAs by means of FTIR spectroscopy and elemental analyses. The elemental analyses of the resulting PAs 5a–f and CoPAs 10a–f were in good agreement with the calculated values for the proposed structure. The representative FTIR spectrum of PA 5f is shown in Figure . The polymer exhibited characteristic absorption bands at 1682 cm−1 for the carbonyl groups (C=O stretching vibration of amidic groups) and 1375 cm−1 (C–N stretching vibration). The absorption bands of amide groups appeared at 3294 cm−1 (N–H stretching). The 1H NMR spectrum of PA 5a showed peaks that confirm its chemical structure, which is shown in Figure the aromatic protons appeared in the region of 7.25–8.11 ppm and the peak in the region of 10.65 ppm is assigned for N–H amide groups in the polymer backbone.
Figure 5 FTIR spectrum of PA 5f.
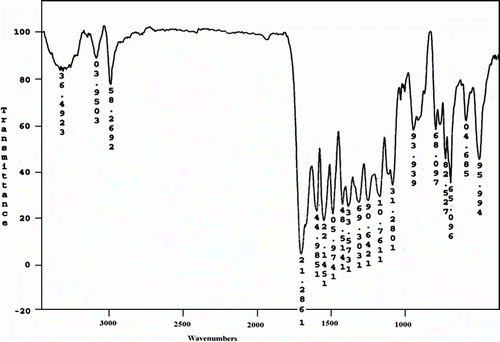
Figure 6 1H NMR spectrum of PA 5a.
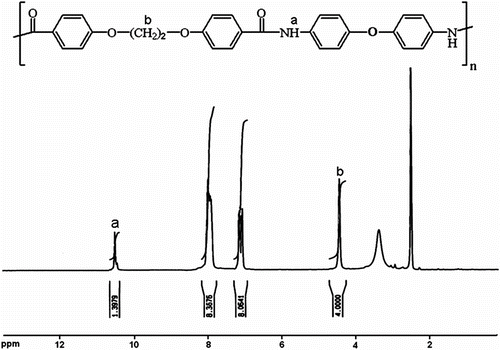
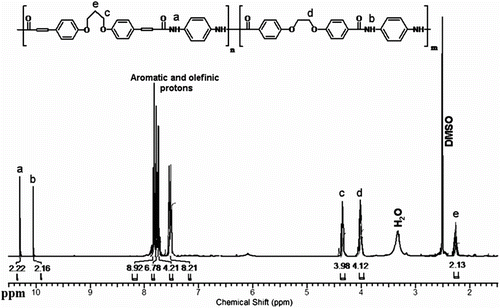
Also, the FTIR spectrum of CoPA 10a is shown in Figure . The polymer exhibited characteristic absorption bands at 1709 cm−1 for the carbonyl groups (C=O stretching vibration of amidic groups) and 1384 cm−1 (C–N stretching vibration). The absorption bands of amide groups appeared at 3340 cm−1 (N–H stretching). The 1H NMR spectrum of CoPA 10d showed peaks that confirm its chemical structure as shown in Figure . The aromatic and olefinic protons appeared in the region of 7.5–7.9 ppm and the two peaks in the regions of 10.5 and 10.3 ppm are assigned for two different N–H amide groups in the polymer backbone.
Figure 7 FTIR spectrum of CoPA 5a.
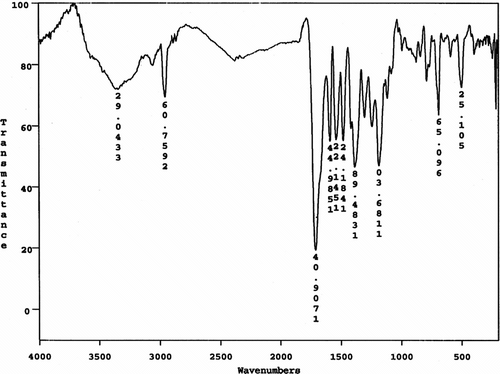
Figure 8 1H NMR spectrum of CoPA 10d.
Solubility of polymers
One of the main objectives of this study was producing modified PAs with improved solubility. The incorporation of monomers with flexible group, such as ether moieties in the polymer backbone, led to these polymers has good solubility in various solvents, especially organic aprotic solvents. The solubility of PAs 5a–f and CoPAs 10a–f was investigated as 0.01 g of polymeric sample in 2 mL of solvent. Remarkably, all of PAs were easily soluble at room temperature in aprotic polar solvents such as NMP, N,N-dimethylacetamide (DMAc), N,N-dimethylformamide (DMF), and tetrahydrofuran (THF), soluble on heating in pyridine (Py) and partially soluble on heating in cyclohexanone, and insoluble in solvents such as acetone, chloroform, ethanol, and methanol. Also, CoPAs were easily soluble at room temperature in aprotic polar solvents such as NMP, DMAc, and DMF, soluble on heating in THF and Py and partially soluble on heating in cyclohexanone, and insoluble in solvents such as acetone, chloroform, ethanol, and methanol (Table ).
Table 2. Solubility of PAs 5a–f and CoPAs 10a–f.
Thermal properties
TGA and derivative of DTG analysis at a rate of 10 °Cmin−1 in a nitrogen atmosphere were utilized to examine the thermal properties of these PAs and CoPAs, and the obtained results are summarized in Table . Figure shows TGA results of PAs 5b, 5d, and 5f and CoPAs 10b, 10d, and 10f.
Table 3. Thermal behavior of PAs 5b, 5d, and 5f and CoPAs 10b, 10d, and 10f.
Figure 9 TGA curves of some PAs and CoPAs.
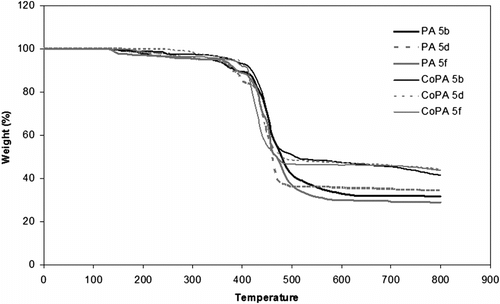
The thermal stability of the polymers was studied on the basis of 5 and 10% weight losses (T 5 and T 10, respectively) of the polymers and the residue at 800 °C (char yield). As given in Table , the range of T 5 and char yield for tree PAs is 340–360 °C and 28.8–34.4%, respectively, but these data for CoPAs are 370–390 °C and 46.3–47.7%, respectively. The TGA data showed that the CoPAs have better thermal properties than PAs. Also, the DSC analyses for PAs and CoPAs showed T g around 184–190 and 192–205 °C, respectively (Table ).
Conclusions
In this article, we have successfully synthesized a new dicarboxylic acid 9, containing ether and olefinic group by a solvent free reaction. Also a series of new organosoluble PAs 5a–f and CoPAs 10a–f were prepared with good thermal properties by the direct polycondensation in TPP/NMP/Py. The results presented herein also clearly demonstrate that incorporating the ether group into the polymer main chain as well as combination of the wholly aromatic backbone and several functional groups remarkably enhanced the solubility in organic solvents of the new polymers, especially about the synthesized PAs. Also, the TGA results showed that these CoPAs have better thermal properties than PAs. These properties could make these PAs and CoPAs attractive for practical applications such as processable high-performance engineering plastics.
Related Research Data
References
- Cassidy , PE . 1980 . Thermally stable polymers , New York , NY : Marcel Dekker .
- Faghihi , KH and Hagibeygi , M . 2003 . New polyamides containing azobenzene unites and hydantoin derivatives in main chain: synthesis and characterization . European Polymer Journal , 39 : 2307 – 14 .
- Mllakpour , S and Kowsari , E . 2005 . Synthesis and characterization of new optically active poly(amide-imide)s containing epiclon and L-methionine moieties in the main chain . Polymers for Advanced Technologies , 16 : 732 – 7 .
- Liaw , DJ , Hsu , PN and Liaw , BY . 2001 . Synthesis and characterization of novel polyamide-imides containing noncoplanar 2,2′-dimethyl-4,4′-biphenylene unit . Journal of Polymer Science Part A: Polymer Chemistry , 39 : 63 – 70 .
- Hajibeygi , M , Shabanian , M and Khodaei-Tehrani , M . 2011 . Synthesis and characterization of new organosoluble poly(amide-imide)s with good thermal properties containing photosensitive and bicyclo segments in the main chain . Designed Monomers and Polymers , 14 : 617 – 33 .
- Nasr Isfahani , H , Faghihi , KH , Hajibeygi , M and Bokaei , M . 2010 . Synthesis and properties of new optically active polyamides containing 1,3-dioxoisoindolin-2-yl units as pendent groups and aromatic diamines . Designed Monomers and Polymers , 13 : 237 – 47 .
- Faghihi , KH and Moghanian , H . 2010 . Synthesis and characterization of new optically active poly(amide-imide)s based on N,N(pyromellitoyl)-bis-L-amino acids and 1,3,4 oxadiazole moieties . Designed Monomers and Polymers , 13 : 207 – 20 .
- Hajibeygi , M , Faghihi , KH and Shabanian , M . 2011 . Synthesis and characterization of novel heat resistance poly(amide-imide)s from N,N0-[2,5-bis(4-aminobenzylidene) cyclopentanone] bistrimellitimide acid and various aromatic diamines . Journal of Applied Polymer Science , 121 : 2877 – 85 .
- Patel , HS , Patel , BP and Patel , DB . 2009 . Synthesis, characterization and glass reinforcement of poly(ester amido imide)s . International Journal of Polymeric Materials , 58 : 625 – 35 .
- Liaw , DJ , Liaw , BY and Yang , CM . 2001 . Synthesis and characterization of new soluble cardo aromatic polyamides bearing diphenylmethylene linkage and norbornyl group . Macromolecular Chemistry and Physics , 202 : 1866 – 72 .
- Ayala , V , Maya , EM , Garcia , JM , de la Campa , JG , Lozano , AE and de Abajo , J . 2005 . Synthesis, characterization, and water sorption properties of new aromatic polyamides containing benzimidazole and ethylene oxide moieties . Journal of Polymer Science Part A: Polymer Chemistry , 43 : 112 – 121 .
- Hsiao , SH , Yang , CP , Chen , CW and Liou , GS . 2005 . Synthesis and properties of novel poly(amide-imide)s containing pendent diphenylamino groups . European Polymer Journal , 41 : 511 – 7 .
- Sava , I and Bruma , M . 2006 . Aromatic polyamides containing pendent acetoxybenzamide groups . Macromolecular Symposium , 239 : 36 – 42 .
- Hale , WF , Farnham , AG , Johnson , RN and Clendinning , RA . 1976 . Poly(aryl ethers) by nucleophilic aromatic substitution. II. Thermal stability . Journal of Polymer Science Part A: Polymer Chemistry , 5 : 2399 – 414 .
- Johnson , RN , Farnham , AG , Clendinning , RA , Hale , WF and Merriman , CN . 1967 . Poly(aryl ethers) by nucleophilic aromatic substitution. I. Synthesis and properties . Journal of Polymer Science Part A: Polymer Chemistry , 5 : 2375 – 98 .
- Faghihi , KH , Shabanian , M and Valikhani , N . 2011 . New poly(amide-imide)s based on 1,3-bis[4,4′-(trimellitimido) phenoxy] propane and hydantoin derivatives: synthesis and properties . Designed Monomers and Polymers , 14 : 109 – 19 .
- Gutch , PK , Banerjee , S and Jaiswal , DK . 2003 . Synthesis and properties of novel aromatic polyamides derived from 2,2-bis[4-(4-amino-2-fluorophenoxy) phenyl]hexafluoropropane and aromatic dicarboxylic acids . Journal of Applied Polymer Science , 89 : 691 – 6 .
- Yamazaki , N , Matsumoto , M and Higashi , F . 1975 . Studies on reactions of the N-phosphonium salts of pyridines. XIV. Wholly aromatic polyamides by the direct polycondensation reaction by using phosphites in the presence of metal salts . Journal of Polymer Science Part A: Polymer Chemistry , 13 : 1373 – 80 .
- Shabanian M. Ghanbari, D. Synthesis of magnesium hydroxide nanofiller and its use for improving thermal properties of new poly(ether-amide). Journal of Applied Polymer Science. doi: 10.1002/app.37640.