
Abstract
A family of polymerizable 2-substituted-1,3-propylidenediphosphonic acids was successfully synthesized for an application in dental adhesives. Methacrylates as well as N-alkyl-acrylamides were prepared. Those monomers were synthesized in 4 to 6 steps and were fully characterized by 1H NMR, 13C NMR, 31P NMR spectroscopy and by HRMS. In order to evaluate their reactivity, the copolymerization of each monomer with N,N′-diethyl-1,3-bis-(acrylamido)propane (DEBAAP) in DMF was investigated by photo-DSC. Bis-(4-methoxybenzoyl)diethylgermanium (BMDG) was added as the photoinitiator to each mixture of acidic monomer/DEBAAP/DMF (2/3/5: mol/mol/mol). It was demonstrated that each monomer efficiently copolymerized with DEBAAP. Self-etching enamel-dentin adhesives containing these new diphosphonic acids were formulated and used to mediate a bond between the dental hard tissues (dentin and enamel) and a restorative material. According to both dentin and enamel shear bond strength measurements, 2-methacryloyloxymethyl-1,3-propylidenediphosphonic acid 8, 2-[N-(2-methacryloyloxyethyl)-carbamoyloxymethyl]-1,3-propylidenediphosphonic acid 10a and 2-[N-(10-methacryloyloxydecyl)-carbamoyloxymethyl]-1,3-propylidenediphosphonic acid 10b were the best candidates to enter new formulations. Those diphosphonic acids were significantly more efficient regarding the etching of enamel than the 1,3-bis(methacrylamido)propane-2-yl dihydrogen phosphate 17, a monomer already used in some commercially available dental formulations.
1. Introduction
Self-etching enamel-dentin adhesives (SEAs) are broadly used in dentistry in order to achieve a strong bond between the dental hard tissues and a dental restoration.[2–4]Citation2Citation3Citation4 Contrary to the total-etch systems, which require the use of a phosphoric acid gel prior to the application of the adhesive, SEAs do not need a separate etching step. SEAs are aqueous solutions containing an acidic monomer (carboxylic acid, dihydrogen phosphate, phosphonic acid) as well as various monofunctional and crosslinking comonomers. The ionization of the acidic monomer is responsible for the demineralization of both dentin and enamel. A major characteristic of SEAs is that both the demineralization of the dental hard tissues and the infiltration of the monomers occur simultaneously. Consequently, the occurrence of post-operative sensitivity is significantly reduced.
Although the micro-mechanical interlocking is essential to reach a good adhesion, the interaction between the acidic monomer and hydroxyapatite (HAP) has been shown to have a great influence on the bond strength.[5–7]Citation5Citation6Citation7 This last parameter being obviously related to the structure of the acidic monomer, a wide range of polymerizable dihydrogen phosphates, phosphonic and bisphosphonic acids were synthesized in order to improve the SEAs performance.[1,2,8–13]Citation1Citation2Citation8Citation9Citation10Citation11Citation12Citation13 Recently, the synthesis of the 3-(methacryloyloxy)-2,2-[di((dihydroxyphosphoryl)methyl)]propyl methacrylate (MDMP) was reported (Figure ).Citation[8] This monomer exhibits great adhesive properties and is a potential candidate to enter new adhesive formulations.
Figure 1 Structure of MDMP.
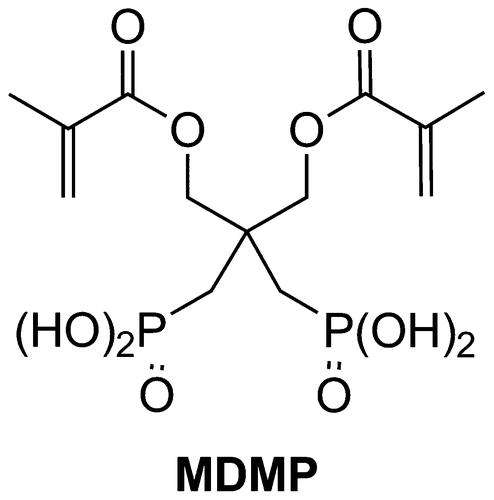
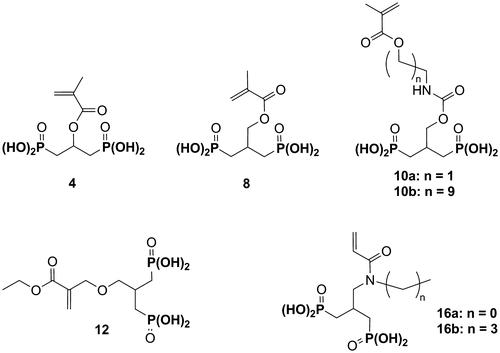
The objective of this study was to extend this work by synthesizing several 2-substituted-1,3-propylidenediphosphonic acids and evaluating them in SEAs (Figure ). In this article, the syntheses, characterization, photopolymerization behaviour and adhesive properties of the monomers 4, 8, 10a–b, 12 and 16a–b are described.
Figure 2 Structure of the monomers 4, 8, 10a,b, 12 and 16a,b.
2 Experimental
2.1 Abbreviations
Acetonitrile (MeCN), Butylated hydroxytoluene (BHT), bis-(4-methoxybenzoyl)diethylgermanium (BMDG), dichloromethane (DCM), double-bond conversion (DBC), N,N′-diethyl-1,3-bis-(acrylamido)propane (DEBAAP), 4-dimethylaminopyridine (DMAP), ethanol (EtOH), ethyl acetate (EA), HAP, 2-hydroxyethyl methacrylate (HEMA), isocyanatoethyl methacrylate (IEM), methanol (MeOH), potassium cyanate (KOCN), pyridinium p-toluenesulfonate (PPTS), rate of polymerization (Rp), shear bond strength (SBS), standard deviation (SD), tetrabutylammonium bromide (NBu4Br), tetramethylsilane (TMS), tetrahydrofuran (THF), triethylamine (TEA), triethylphosphite (TEP), trimethylsilyl bromide (TMSBr), zinc chloride (ZnCl2).
2.2 Materials
Excepted for the preparation of the compound 5, all reactions were carried out under an argon atmosphere. DCM, MeOH, MeCN and THF were dried over molecular sieves. TMSBr was distilled prior to use. KOCN and NBu4Br were dried at 100 °C under vacuum (0.1 mbar for KOCN and 3 mbar for NBu4Br) for 4 h prior to use. IEM was purchased from ABCR (Germany). Ethyl 2-(chloromethyl)acrylate was purchased from Synthon Chemical (Germany). All other reagents were purchased from Sigma–Aldrich (Switzerland) and were used without further purification. Diethyl 2,3-epoxypropylphosphonate 1,Citation[14] tetraethyl 2-methylene-1,3-propylendiphosphonate 5 Citation[15] and tetraethyl 2-hydroxymethyl-1,3-propylenediphosphonate 6 Citation[16] were prepared according to the literature. Column chromatographies were performed on Macherey-Nagel silica gel 60 (40–63 μm). Thin layer chromatography (TLC) was performed on silica gel 60 F-254 plates.
2.3 Methods
1H NMR, 13C NMR and 31P NMR spectra were recorded on a DPX-400 spectrometer using TMS as internal reference for 1H NMR and 13C NMR chemical shifts and using H3PO4 (85%) as external reference for 31P NMR chemical shifts. Data are given in the following order: chemical shift in ppm, multiplicity (s, singlet; d, doublet; t, triplet; q, quadruplet; qt, quintuplet; m, multiplet), coupling constant in Hertz, assignment. High-resolution mass spectra (HRMS) were obtained with a Waters Q-TOF Micro instrument in electrospray ionization positive (ES+) mode and lockspray with orthophosphoric acid. These analyses were performed with an infusion introduction of 10 μL/min, a source temperature of 80 °C, a desolvatation temperature of 120 °C and an external calibration with NaI. The melting points (Mp) were measured with a Perkin Elmer differential scanning calorimeter (DSC), Pyris Diamond.
2.4 Syntheses
2.4.1 Synthesis of the acidic monomer 4
2.4.1.1 Tetraethyl 2-hydroxy-1,3-propylidenediphosphonate 2
TEP (50.2 mL, 0.29 mol, 5.0 eq.) was added to ZnCl2 (8.37 g, 0.061 mol, 1.05 eq.). Diethyl 2,3-epoxypropylphosphonate (11.35 g, 0.058 mol) was then added dropwise and the solution was stirred for 15 h at room temperature. EA (250 mL) was added and the solution was washed twice with an ammonium chloride saturated aqueous solution (2 × 125 mL). The combined aqueous layers were extracted twice with EA (2 × 125 mL). The combined organic layers were dried over anhydrous sodium sulfate and concentrated under reduced pressure. The excess of TEP was recovered by distillation under reduced pressure (P: ca. 17 mm Hg, T: 70 °C). The product was heated at 100 °C under high vacuum (0.08 mbar). The crude product was finally purified by flash column chromatography (Eluent: EA/MeOH: 9/1). 9.3 g (0.028 mol) of the desired compound were isolated.
Yield: 48%. Aspect: slightly yellow oil. 1H NMR (400 MHz, CDCl3): δ = 1.31, 1.32 (2t, 3JHH = 7.0 Hz, 12H, POCH2CH3); 2.00–2.22 (m, 4H, CH2P); 4.04–4.19 (m, 9H, POCH2CH3 and OH); 4.30–4.45 (m, 1H, CHOH). 31P NMR (162 MHz, CDCl3): δ = 28.4. 13C NMR (101 MHz, CDCl3): δ = 16.3–16.5 (m, POCH2CH3); 34.2 (dd, 1JCp = 139.3 Hz, 3JCp = 13.2 Hz, CH2P); 61.8–62.2 (m, POCH2CH3); 62.4 (t, 2JCp = 2.9 Hz, CHOH).
2.4.1.2 Tetraethyl 2-methacryloyloxy-1,3-propylidenediphosphonate 3
Methacrylic anhydride (4.0 mL, 27.0 mmol, 1.5 eq.) was added, under stirring, to a solution of the hydroxyphosphonate 2 (5.97 g, 18.0 mmol), TEA (3.76 mL, 27.0 mmol, 1.5 eq.) and DMAP (176 mg, 1.4 mmol, 8 mol%) in anhydrous DCM (50 mL). The mixture was stirred for 40 h under reflux. Deionised water (25 mL) was added. The layers were separated and the aqueous layer was extracted twice with DCM (2 × 25 mL). The combined organic layers were dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by flash column chromatography (eluent: EA/MeOH: 9/1). 6.1 g (15.2 mmol) of the desired compound were isolated.
Yield: 85%. Aspect: colorless oil. 1H NMR (400 MHz, CDCl3): δ = 1.29, 1.31 (2t, 3JHH = 7.1 Hz, 12H, POCH2CH3); 1.94 (s, 3H, CH3); 2.26–2.49 (m, 4H, CH2P); 4.03–4.16 (m, 8H, POCH2CH3); 5.33–5.46 (m, 1H, CHO); 5.58–5.62 (m, 1H, CH2=C); 6.18 (ls, 1H, CH2=C). 31P NMR (162 MHz, CDCl3): δ = 25.7. 13C NMR (101 MHz, CDCl3): δ = 16.3 (d, 3JCp = 6.4 Hz, POCH2CH3); 16.4 (d, 3JCp = 6.4 Hz, POCH2CH3); 18.2 (s, CH3); 31.1 (dd, 1JCp = 139.2 Hz, 3JCp = 8.1 Hz, CH2P); 61.8 (d, 2JCp = 6.6 Hz, POCH2CH3); 61.9 (d, 2JCp = 6.6 Hz, POCH2CH3); 65.7 (t, 2JCp = 2.2 Hz, CHO); 126.3 (s, CH2=C); 136.0 (s, CH2=C); 166.2 (s, C=O).
2.4.1.3 2-Methacryloyloxy-1,3-propylidenediphosphonic acid 4
TMSBr (8.8 mL, 66.6 mmol, 6.0 eq.) was added to a solution of the diphosphonate 3 (4.44 g, 11.1 mmol) in anhydrous DCM (22 mL). After stirring for 5 h at 30 °C, the mixture was concentrated under reduced pressure. MeOH (25 mL) was added and the mixture was stirred for 30 min at room temperature. BHT (200 ppm) and galvinoxyl free radical (20 ppm) were added to the solution. The solvent was evaporated and the product was dried to a constant weight under vacuum (0.1 mbar). 3.10 g (10.8 mmol) of the desired compound were isolated.
Yield: 97%. Aspect: slightly orange highly viscous oil. 1H NMR (400 MHz, D2O): δ = 1.77 (s, 3H, CH3); 2.12–2.30 (m, 4H, CH2P); 5.27–5.41 (m, 1H, CHO); 5.54–5.59 (m, 1H, CH2=C); 6.00 (ls, 1H, CH2=C). 31P NMR (162 MHz, D2O): δ = 23.9. 13C NMR (101 MHz, D2O): δ = 17.2 (s, CH3); 32.3 (dd, 1JCp = 135.1 Hz, 3JCp = 10.8 Hz, CH2P); 65.7 (t, 2JCp = 3.7 Hz, CHO); 127.0 (s, CH2=C); 135.7 (s, CH2=C); 168.1 (s, C=O). HRMS (m/z): calcd for C7H15O8P2: 289.0242; found: 289.0239 [M + H]+.
2.4.2 Synthesis of the acidic monomer 8
2.4.2.1 Tetraethyl 2-methacryloyloxymethyl-1,3-propylidenediphosphonate 7
Methacrylic anhydride (1.45 mL, 9.7 mmol, 1.5 eq.) was added, under stirring, to a solution of the hydroxyphosphonate 6 (2.24 g, 6.5 mmol), TEA (1.35 mL, 9.7 mmol, 1.5 eq.) and DMAP (63 mg, 0.5 mmol, 8 mol%) in anhydrous DCM (15 mL). The mixture was stirred for 6 h at RT. Deionised water (15 mL) was added. The layers were separated and the aqueous layer was extracted twice with EA (2 × 15 mL). The combined organic layers were dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by flash column chromatography (eluent: EA/MeOH: 91/9). 2.0 g (4.8 mmol) of the desired compound were isolated.
Yield: 75%. Aspect: colorless oil. 1H NMR (400 MHz, CDCl3): δ = 1.31, 1.32 (2t, 3JHH = 7.1 Hz, 12H, POCH2CH3); 1.90–2.16 (m, 4H, CH2P); 1.94 (s, 3H, CH3); 2.53–2.69 (m, 1H, CHCH2P); 4.04–4.17 (m, 8H, POCH2CH3); 4.27 (d, 3JHH = 5.1 Hz, 2H, CH2OCO); 5.56–5.59 (m, 1H, CH2=C); 6.10 (ls, 1H, CH2=C). 31P NMR (162 MHz, CDCl3): δ = 29.3. 13C NMR (101 MHz, CDCl3): δ = 16.4 (d, 3JCp = 6.3 Hz, POCH2CH3); 18.4 (s, CH3); 27.9 (dd, 1JCp = 141.1 Hz, 3JCp = 10.5 Hz, CH2P); 28.9 (t, 2JCp = 3.9 Hz, CHCH2P); 61.7 (d, 2JCp = 7.7 Hz, POCH2CH3); 66.9 (t, 3JCp = 9.2 Hz, CH2OCO); 125.8 (s, CH2=C); 136.1 (s, CH2=C); 166.9 (s, C=O).
2.4.2.2 2-Methacryloyloxymethyl-1,3-propylidenediphosphonic acid 8
The monomer 8 was prepared from the diphosphonate 7 (1.86 g, 4.5 mmol) according to the procedure described for the synthesis of the monomer 4. 1.36 g (4.5 mmol) of the desired compound were isolated.
Yield: 100%. Aspect: white paste. 1H NMR (400 MHz, D2O): δ = 1.89 (s, 3H, CH3); 1.99 (dd, 3JHH = 6.8 Hz, 2JHp = 18.1 Hz, 4H, CH2P); 2.43–2.58 (m, 1H, CHCH2P); 4.23 (d, 3JHH = 5.0 Hz, 2H, CH2OCO); 5.65–5.68 (m, 1H, CH2=C); 6.11 (ls, 1H, CH2=C). 31P NMR (162 MHz, D2O): δ = 29.9. 13C NMR (101 MHz, D2O): δ = 17.5 (s, CH3); 28.8 (t, 2JCp = 3.8 Hz, CHCH2P); 29.3 (dd, 1JCp = 135.6 Hz, 3JCp = 10.7 Hz, CH2P); 67.4 (t, 3JCp = 8.8 Hz, CH2OCO); 127.1 (s, CH2=C); 135.9 (s, CH2=C); 169.8 (s, C=O). HRMS (m/z): calcd for C8H17O8P2: 303.0399; found: 303.0403 [M + H]+.
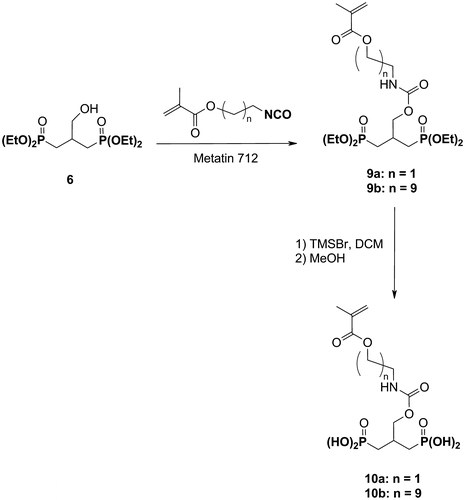
2.4.3 Synthesis of the acidic monomer 10a
2.4.3.1 Tetraethyl 2-[N-(2-methacryloyloxyethyl)-carbamoyloxymethyl]-1,3-propylidenediphosphonate 9a
A 1% solution of dibutyltin dilaurate in anhydrous DCM (4.0 mL) was added to a solution of the hydroxyphosphonate 6 (4.8 g, 13.8 mmol) in anhydrous DCM (6.0 mL). IEM (1.96 mL, 13.8 mmol) was subsequently added dropwise to the mixture. The solution was stirred for 3 h at RT and concentrated under reduced pressure. The crude product was purified by flash column chromatography (eluent: EA/MeOH: 95/5). 6.0 g (12.0 mmol) of the desired compound were isolated.
Yield: 86%. Aspect: colorless oil. 1H NMR (400 MHz, CDCl3): δ = 1.31, 1.32 (2t, 3JHH = 7.1 Hz, 12H, POCH2CH3); 1.85–2.12 (m, 4H, CH2P); 1.94 (s, 3H, CH3); 2.43–2.62 (m, 1H, CHCH2P); 3.48 (dt, 3JHH = 5.4 Hz, 3JHH = 5.4 Hz, 2H, CH2NH); 4.03–4.16 (m, 8H, POCH2CH3); 4.19 (d, 3JHH = 5.2 Hz, 2H, CH2OCONH); 4.22 (t, 3JHH = 5.4 Hz, 2H, NHCH2CH2O); 5.07 (t, 3JHH = 5.4 Hz, 1H, NH); 5.58–5.62 (m, 1H, CH2=C); 6.12 (ls, 1H, CH2=C). 31P NMR (162 MHz, CDCl3): δ = 29.5. 13C NMR (101 MHz, CDCl3): δ = 16.4 (d, 3JCp = 6.1 Hz, POCH2CH3); 18.3 (s, CH3); 27.8 (dd, 1JCp = 141.2 Hz, 3JCp = 10.2 Hz, CH2P); 29.1 (t, 2JCp = 3.9 Hz, CHCH2P); 40.2 (s, CH2NH); 61.7 (2d, 2JCp = 6.8 Hz, POCH2CH3); 63.7 (s, NHCH2CH2O); 67.3 (t, 3JCp = 9.4 Hz, CH2OCONH); 126.1 (s, CH2=C); 136.0 (s, CH2=C); 156.2 (s, NHCO); 167.3 (s, C=O).
2.4.3.2. 2-[N-(2-Methacryloyloxyethyl)-carbamoyloxymethyl]-1,3-propylidenediphosphonic acid 10a
The monomer 10a was prepared from the diphosphonate 9a (4.31 g, 8.6 mmol) according to the procedure described for the synthesis of the monomer 4. 3.11 g (8.0 mmol) of the desired compound were isolated.
Yield: 93%. Aspect: highly viscous oil. 1H NMR (400 MHz, MeOD): δ = 1.90–2.10 (m, 4H, CH2P); 1.93 (s, 3H, CH3); 2.42–2.62 (m, 1H, CHCH2P); 3.40 (t, 3JHH = 5.4 Hz, 2H, CH2NH); 4.14–4.22 (m, 4H, CH2OCONH and NHCH2CH2O); 5.63 (ls, 1H, CH2=C); 6.12 (ls, 1H, CH2=C). 31P NMR (162 MHz, MeOD): δ = 28.1. 13C NMR (101 MHz, MeOD): δ = 18.5 (s, CH3); 30.3 (dd, 1JCp = 138.1 Hz, 3JCp = 9.7 Hz, CH2P); 30.8 (t, 2JCp = 3.3 Hz, CHCH2P); 40.8 (s, CH2NH); 64.7 (s, NHCH2CH2O); 68.5 (t, 3JCp = 9.8 Hz, CH2OCONH); 126.6 (s, CH2=C); 137.6 (s, CH2=C); 158.9 (s, NHCO); 168.8 (s, C=O). HRMS (m/z): calcd for C11H22NO10P2: 390.0719; found: 390.0722 [M + H]+.
2.4.4 Synthesis of the acidic monomer 10b
2.4.4.1 10-Bromodecyl methacrylate
2,4,6-Collidine (12.3 mL, 92.7 mmol, 1.1 eq.) was added to a solution of 10-bromodecanol (20.0 g, 84.3 mmol) in anhydrous DCM (200 mL). The mixture was cooled down to 0 °C and methacryloyl chloride (16.5 mL, 168.6 mmol, 2.0 eq.) was slowly added. The solution was stirred for 1 h at 0 °C and 15 h at RT. The solution was washed with deionized water (100 mL) and with a cold HCl 1 N solution (100 mL). The organic layer was dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by flash column chromatography (eluent: Hexane/DCM: 8/2). 18.0 g (59.0 mmol) of the desired compound were isolated.
Yield: 70%. Aspect: colorless liquid. 1H NMR (400 MHz, CDCl3): δ = 1.26–1.47 (m, 12H, CH2); 1.67 (qt, 3JHH = 7.5 Hz, 2H, CH2); 1.85 (qt, 3JHH = 7.5 Hz, 2H, CH2); 1.94 (s, 3H, CH3); 3.40 (t, 3JHH = 6.8 Hz, 2H, CH2Br); 4.14 (t, 3JHH = 6.7 Hz, 2H, CH2O); 5.53–5.56 (m, 1H, CH2=C); 6.01 (ls, 1H, CH2=C). 13C NMR (101 MHz, CDCl3): δ = 18.3 (s, CH3); 25.9 (s, CH2); 28.1 (s, CH2); 28.6 (s, CH2); 28.7 (s, CH2); 29.2 (s, CH2); 29.3 (s, CH2); 29.4 (s, CH2); 32.8 (s, CH2); 33.9 (s, CH2Br); 64.7 (s, CH2O); 125.1 (s, CH2=C); 136.5 (s, CH2=C); 167.4 (s, C=O).
2.4.4.2 Tetraethyl 2-[N-(10-methacryloyloxydecyl)-carbamoyloxymethyl]-1,3-propylidenediphosphonate 9b
A solution of 10-bromodecyl methacrylate (8.36 g, 27.3 mmol) diluted in anhydrous MeCN (55 mL) was added to a slurry of KOCN (3.33 g, 41.0 mmol) and NBu4Br (1.77 g, 5.48 mmol). The mixture was stirred for 64 h. The solution was concentrated under reduced pressure and hexane (100 mL) was added. The solution was filtrated and concentrated under reduced pressure. 4.75 g of a yellow oil were obtained. A solution of the hydroxyphosphonate 6 (2.41 g, 7.0 mmol) in anhydrous DCM (2.0 mL) was added to the crude product. A 1% solution of dibutyltin dilaurate in anhydrous DCM (2.0 mL) was subsequently introduced. The solution was stirred for 15 h at RT and concentrated under reduced pressure. The crude product was purified by flash column chromatography (eluent: EA/MeOH: 95/5). 3.85 g (6.3 mmol) of the desired compound were isolated.
Yield: 90%. Aspect: colorless oil. 1H NMR (400 MHz, CDCl3): δ = 1.23–1.40 (m, 12H, CH2); 1.31, 1.32 (2t, 3JHH = 7.1 Hz, 12H, POCH2CH3); 1.42–1.53 (m, 2H, CH2); 1.66 (qt, 3JHH = 6.7 Hz, 2H, CH2); 1.85–2.12 (m, 4H, CH2P); 1.94 (s, 3H, CH3); 2.42–2.62 (m, 1H, CHCH2P); 3.14 (dt, 3JHH = 6.6 Hz, 3JHH = 6.6 Hz, 2H, CH2NH); 4.03–4.15 (m, 10H, POCH2CH3 and CH2CH2O); 4.16 (d, 3JHH = 5.2 Hz, 2H, CH2OCONH); 4.73 (t, 3JHH = 6.6 Hz, 1H, NH); 5.52–5.55 (m, 1H, CH2=C); 6.09 (ls, 1H, CH2=C). 31P NMR (162 MHz, CDCl3): δ = 29.6. 13C NMR (101 MHz, CDCl3): δ = 16.4 (d, 3JCp = 6.1 Hz, POCH2CH3); 18.3 (s, CH3); 26.0 (s, CH2); 26.8 (s, CH2); 27.8 (dd, 1JCp = 140.8 Hz, 3JCp = 10.0 Hz, CH2P); 28.6 (s, CH2); 29.2 (t, 2JCp = 4.1 Hz, CHCH2P); 29.2 (s, CH2); 29.3 (2s, 2CH2); 29.4 (s, CH2); 30.0 (s, CH2); 41.1 (s, CH2NH); 61.7 (2d, 2JCp = 6.4 Hz, POCH2CH3); 64.8 (s, CH2CH2O); 67.0 (t, 3JCp = 9.4 Hz, CH2OCONH); 125.2 (s, CH2=C); 136.7 (s, CH2=C); 156.3 (s, NHCO); 167.6 (s, C=O).
2.4.4.3 2-[N-(10-Methacryloyloxydecyl)-carbamoyloxymethyl]-1,3-propylidenediphosphonic acid 10b
The monomer 10b was prepared from the diphosphonate 9b (5.06 g, 8.3 mmol) according to the procedure described for the synthesis of the monomer 4. 3.97 g (7.9 mmol) of the desired compound were isolated.
Yield: 96%. Aspect: slightly orange solid. Mp: 59 °C. 1H NMR (400 MHz, MeOD): δ = 1.28–1.43 (m, 12H, CH2); 1.44–1.54 (m, 2H, CH2); 1.67 (qt, 3JHH = 6.9 Hz, 2H, CH2); 1.92 (s, 3H, CH3); 1.94–2.10 (m, 4H, CH2P); 2.43–2.61 (m, 1H, CHCH2P); 3.08 (t, 3JHH = 7.1 Hz, 2H, CH2NH); 4.13 (t, 3JHH = 6.6 Hz, 2H, CH2CH2O); 4.17 (d, 3JHH = 5.4 Hz, 2H, CH2OCONH); 5.58–5.62 (m, 1H, CH2=C); 6.07 (ls, 1H, CH2=C). 31P NMR (162 MHz, MeOD): δ = 27.9. 13C NMR (101 MHz, MeOD): δ = 18.4 (s, CH3); 27.1 (s, CH2); 27.9 (s, CH2); 29.7 (s, CH2); 30.3 (dd, 1JCp = 137.2 Hz, 3JCp = 10.0 Hz, CH2P); 30.3 (s, CH2); 30.4 (s, CH2); 30.6 (2s, 2CH2); 30.8 (t, 2JCp = 3.5 Hz, CHCH2P); 31.0 (s, CH2); 41.9 (s, CH2NH); 66.0 (s, CH2CH2O); 68.5 (t, 3JCp = 9.3 Hz, CH2OCONH); 126.0 (s, CH2=C); 137.9 (s, CH2=C); 158.9 (s, NHCO); 168.9 (s, C=O). HRMS (m/z): calcd for C19H38NO10P2: 502.1971; found: 502.1952 [M + H]+.
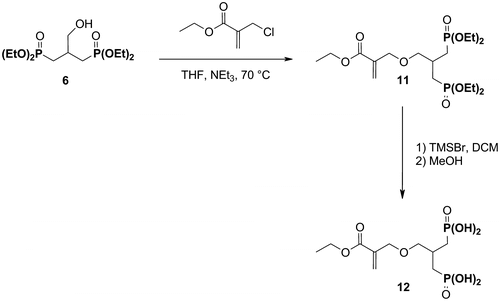
2.4.5 Synthesis of the acidic monomer 12
2.4.5.1 Ethyl 2-[2-oxa-4,4-(di((diethoxyphosphoryl)methyl)butyl]acrylate 11
Ethyl 2-(chloromethyl)acrylate (3.68 g, 26.0 mmol, 1.5 eq.) was added to a solution of the hydroxyphosphonate 6 (6.0 g, 17.0 mmol) and TEA (3.62 mL, 26 mmol) in anhydrous THF (30 mL). The reaction was stirred for 45 h at 70 °C and was concentrated under reduced pressure. Deionized water (50 mL) was added and the solution was extracted with EA (3 × 50 mL). The organic phases were combined and dried over anhydrous sodium sulfate. After filtration, the solution was concentrated under reduced pressure. The crude product was purified by flash column chromatography (eluent: EA/MeOH: 98/2). 3.77 g (8.2 mmol) of the desired compound were isolated.
Yield: 48%. Aspect: slightly yellow oil. 1H NMR (400 MHz, CDCl3): δ = 1.28 (t, 3JHH = 7.9 Hz, 3H, COOCH2CH3); 1.30 (t, 3JHH = 7.8 Hz, 12H, POCH2CH3); 1.92–2.10 (m, 4H, CH2P); 2.37–2.53 (m, 1H, CHCH2O); 3.60 (d, 3JHH = 5.1 Hz, 2H, CHCH2O); 4.00–4.15 (m, 8H, POCH2CH3); 4.17 (s, 2H, OCH2C); 4.20 (q, 3JHH = 7.1 Hz, 2H, COOCH2CH3); 5.81 (s, 1H, CH2=C); 6.26 (s, 1H, CH2=C). 31P NMR (162 MHz, CDCl3): δ = 30.3. 13C NMR (101 MHz, CDCl3): δ = 14.2 (s, COOCH2CH3); 16.4 (d, 3JCp = 5.7 Hz, POCH2CH3); 27.8 (dd, 1JCp = 140.2 Hz, 3JCp = 10.7 Hz, CH2P); 29.7 (t, 2JCp = 3.7 Hz, CHCH2O); 60.7 (s, COOCH2CH3); 61.5 (2d, 2JCp = 6.6 Hz, POCH2CH3); 69.1 (s, OCH2C); 73.1 (t, 3JCp = 8.6 Hz, CHCH2O); 125.4 (s, CH2=C); 137.4 (s, CH2=C); 165.8 (s, C=O).
2.4.5.2 Ethyl 2-[2-oxa-4,4-(di((dihydroxyphosphoryl)methyl)butyl]acrylate 12
The monomer 12 was prepared from the diphosphonate 11 (3.65 g, 8.0 mmol) according to the procedure described for the synthesis of the monomer 4. 2.76 g (8.0 mmol) of the desired compound were isolated.
Yield: 100%. Aspect: highly viscous oil. 1H NMR (400 MHz, D2O): δ = 1.27 (t, 3JHH = 7.1 Hz, 3H, COOCH2CH3); 1.87–2.04 (m, 4H, CH2P); 2.30–2.45 (m, 1H, CHCH2O); 3.59 (d, 3JHH = 5.7 Hz, 2H, CHCH2O); 4.23 (q, 3JHH = 7.1 Hz, 2H, COOCH2CH3); 4.25 (s, 2H, OCH2C); 5.94 (s, 1H, CH2=C); 6.35 (s, 1H, CH2=C). 31P NMR (162 MHz, D2O): δ = 28.4. 13C NMR (101 MHz, D2O): δ = 13.3 (s, COOCH2CH3); 28.8 (dd, 1JCp = 134.5 Hz, 3JCp = 9.6 Hz, CH2P); 29.1 (t, 2JCp = 2.9 Hz, CHCH2O); 62.0 (s, COOCH2CH3); 69.3 (s, OCH2C); 72.6 (t, 3JCp = 9.2 Hz, CHCH2O); 129.6 (s, CH2=C); 136.2 (s, CH2=C); 168.1 (s, C=O). HRMS (m/z): calcd for C10H21O9P2: 347.0661; found: 347.0664 [M + H]+.
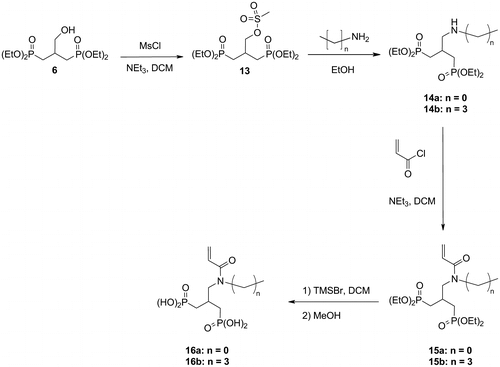
2.4.6 Synthesis of the acidic monomers 16a and 16b
2.4.6.1 Tetraethyl 2-methanesulfonyloxymethyl-1,3-propylidenediphosphonate 13
Methanesulfonyl chloride (2.2 mL, 28.6 mmol, 1.1 eq.) was added dropwise, at 0 °C, to a solution of the hydroxyphosphonate 6 (9.0 g, 26.0 mmol) and TEA (4.0 mL, 28.6 mmol, 1.1 eq.) in anhydrous DCM (90 mL). The mixture was stirred for 30 min at 0 °C and 2 h at RT. The solution was concentrated under reduced pressure. Deionized water (100 mL) was added and the solution was extracted with EA (3 × 100 mL). The combined organic layers were dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by flash column chromatography (eluent: EA/MeOH: 9/1). 9.37 g (22.1 mmol) of the desired compound were isolated.
Yield: 85%. Aspect: colorless oil. 1H NMR (400 MHz, CDCl3): δ = 1.33 (t, 3JHH = 7.1 Hz, 12H, POCH2CH3); 1.89–2.14 (m, 4H, CH2P); 2.51–2.69 (m, 1H, CHCH2P); 3.06 (s, 3H, CH3S); 4.04–4.16 (m, 8H, POCH2CH3); 4.42 (d, 3JHH = 4.5 Hz, 2H, CH2OS). 31P NMR (162 MHz, CDCl3): δ = 28.4. 13C NMR (101 MHz, CDCl3): δ = 16.4 (d, 3JCp = 6.3 Hz, POCH2CH3); 27.5 (dd, 1JCp = 141.0 Hz, 3JCp = 11.3 Hz, CH2P); 29.3 (t, 2JCp = 3.9 Hz, CHCH2P); 37.1 (s, CH3S); 61.8, 61.9 (2d, 2JCp = 6.5 Hz, POCH2CH3); 72.1 (t, 3JCp = 8.1 Hz, CH2OS).
2.4.6.2 Tetraethyl 2-[(N-methyl)aminomethyl]-1,3-propylidenediphosphonate 14a
A solution of the mesylate 13 (9.7 g, 22.9 mmol) diluted in dry EtOH (60 mL) was added to a 33 wt.% solution of methylamine in EtOH (57 mL). The mixture was stirred for 50 h at 40 °C. The solution was concentrated under reduced pressure and deionized water (30 mL) was added. A 10 wt.% NaOH aqueous solution was added up to pH > 11. The solution was extracted with DCM (3 × 50 mL). The combined organic layers were dried over anhydrous sodium sulfate and concentrated under reduced pressure. 7.47 g (20.8 mmol) of the desired compound were isolated. The aminophosphonate 14a was used in the next step without further purification.
Yield: 91%. Aspect: slightly yellow oil. 1H NMR (400 MHz, CDCl3): δ = 1.31 (t, 3JHH = 7.0 Hz, 12H, POCH2CH3); 1.91–2.09 (m, 4H, CH2P); 2.25–2.41 (m, 1H, CHCH2P); 2.40 (s, 3H, CH3N); 2.71 (d, 3JHH = 6.0 Hz, 2H, CHCH2N); 4.01–4.16 (m, 8H, POCH2CH3). 31P NMR (162 MHz, CDCl3): δ = 30.7. 13C NMR (101 MHz, CDCl3): δ = 16.4 (d, 3JCp = 5.9 Hz, POCH2CH3); 28.7 (dd, 1JCp = 139.5 Hz, 3JCp = 10.4 Hz, CH2P); 29.2 (t, 2JCp = 3.5 Hz, CHCH2P); 36.5 (s, CH3N); 56.1 (t, 3JCp = 9.0 Hz, CHCH2N); 61.4, 61.5 (2d, 2JCp = 6.6 Hz, POCH2CH3).
2.4.6.3 Tetraethyl 2-[(N-butyl)aminomethyl]-1,3-propylidenediphosphonate 14b
The mesylate 13 (8.85 g, 20.9 mmol) was added to a solution of butylamine (20.6 mL, 0.21 mol, 10.0 eq.) in EtOH (110 mL). The mixture was stirred for 50 h at 60 °C. The solution was concentrated under reduced pressure and deionized water (50 mL) was added. A 10 wt.% NaOH aqueous solution was added up to pH > 11. The solution was extracted with DCM (3 × 60 mL). The combined organic layers were dried over anhydrous sodium sulfate and concentrated under reduced pressure. 7.0 g (17.5 mmol) of the desired compound were isolated. The aminophosphonate 14b was used in the next step without further purification.
Yield: 84%. Aspect: yellow oil. 1H NMR (400 MHz, CDCl3): δ = 0.90 (t, 3JHH = 7.3 Hz, 3H, CH2CH2CH3); 1.27–1.37 (m, 2H, CH2); 1.32 (t, 3JHH = 7.0 Hz, 12H, POCH2CH3); 1.37–1.48 (m, 2H, CH2); 1.94–2.10 (m, 4H, CH2P); 2.22–2.39 (m, 1H, CHCH2P); 2.57 (t, 3JHH = 7.2 Hz, 2H, CH2CH2N); 2.74 (d, 3JHH = 6.1 Hz, 2H, CHCH2N); 4.01–4.17 (m, 8H, POCH2CH3). 31P NMR (162 MHz, CDCl3): δ = 30.9. 13C NMR (101 MHz, CDCl3): δ = 14.0 (s, CH2CH2CH3); 16.4 (d, 3JCp = 5.4 Hz, POCH2CH3); 20.5 (s, CH2); 28.6 (dd, 1JCp = 139.0 Hz, 3JCp = 9.6 Hz, CH2P); 29.5 (t, 2JCp = 3.9 Hz, CHCH2P); 32.4 (s, CH2); 49.7 (s, CH2CH2N); 53.9 (t, 3JCp = 8.8 Hz, CHCH2N); 61.4, 61.5 (2d, 2JCp = 6.5 Hz, POCH2CH3).
2.4.6.4 Tetraethyl 2-[(N-methyl)acrylamidomethyl]-1,3-propylidenediphosphonate 15a
The aminophosphonate 14a (7.36 g, 20.5 mmol) and TEA (3.14 mL, 22.6 mmol, 1.1 eq.) were diluted in dry DCM (50 mL). The mixture was cooled down to 0 °C. A solution of acryloyl chloride (1.66 mL, 20.5 mmol, 1.0 eq.) diluted in dry DCM (25 mL) was added dropwise to the reaction mixture. The solution was stirred for 30 min at 0 °C and for 2 h at RT. The solution was washed with a 10 wt.% NaOH aqueous solution (2 × 25 mL). The combined aqueous layers were extracted with EA (3 × 50 mL). The combined organic layers were dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by flash column chromatography (eluent: EA/MeOH: 9/1). 6.86 g (16.6 mmol) of the desired compound were isolated.
Yield: 81%. Aspect: yellow oil. 1H NMR (400 MHz, CDCl3): 2 rotamers δ = 1.30, 1.31 (2t, 3JHH = 7.0 Hz, 12H, POCH2CH3); 1.73–1.90 (m, 2H, CH2P); 1.98–2.13 (m, 2H, CH2P); 2.40–2.56 (m, 1H, CHCH2P); 3.01, 3.08 (2s, 3H, CH3N); 3.59, 3.55 (2d, 3JHH = 7.4 Hz, 2H, CHCH2N); 4.01–4.16 (m, 8H, POCH2CH3); 5.69, 5.70 (2dd, 2JHH = 2.0 Hz, 3JHH = 10.4 Hz, 1H, CH2=CH); 6.31, 6.36 (2dd, 2JHH = 2.0 Hz, 3JHH = 16.7 Hz, 1H, CH2=CH); 6.58, 6.67 (2dd, 3JHH = 10.4 Hz, 3JHH = 16.7 Hz, 1H, CH2=CH). 31P NMR (162 MHz, CDCl3): 2 rotamers δ = 28.4, 30.1. 13C NMR (101 MHz, CDCl3): 2 rotamers δ = 16.4 (d, 3JCp = 6.2 Hz, POCH2CH3); 27.7 (dd, 1JCp = 139.9 Hz, 3JCp = 9.0 Hz, CH2P); 27.7 (t, 2JCp = 4.2 Hz, CHCH2P); 28.8 (dd, 1JCp = 139.9 Hz, 3JCp = 9.0 Hz, CH2P); 29.1 (t, 2JCp = 4.2 Hz, CHCH2P); 34.7, 35.4 (2s, CH3N); 51.8, 54.0 (2t, 3JCp = 9.2 Hz, CHCH2N); 61.6, 61.7, 61.8, 61.9 (4d, 2JCp = 6.6 Hz, POCH2CH3); 127.4, 127.6 (2s, CH=CH2); 128.3, 128.5 (2s, CH=CH2); 166.9, 167.1 (2s, C=O).
2.4.6.5 Tetraethyl 2-[(N-butyl)acrylamidomethyl]-1,3-propylidenediphosphonate 15b
The aminophosphonate 14b (6.93 g, 17.3 mmol) and TEA (2.65 mL, 19.0 mmol, 1.1 eq.) were diluted in dry DCM (40 mL). The mixture was cooled down to 0 °C. A solution of acryloyl chloride (1.41 mL, 17.3 mmol, 1.0 eq.) diluted in dry DCM (25 mL) was added dropwise to the reaction mixture. The solution was stirred for 30 min at 0 °C and for 2 h at RT. The solution was washed with a 10 wt.% NaOH aqueous solution (2 × 25 mL). The combined aqueous layers were extracted with EA (3 × 50 mL). The combined organic layers were dried over anhydrous sodium sulfate and concentrated under reduced pressure. The crude product was purified by flash column chromatography (eluent: EA/MeOH: 9/1). 7.0 g (15.4 mmol) of the desired compound were isolated.
Yield: 89%. Aspect: yellow oil. 1H NMR (400 MHz, CDCl3): 2 rotamers δ = 0.91, 0.93 (2t, 3JHH = 7.2 Hz, 3H, CH2CH2CH3); 1.26–1.36 (m, 2H, CH2); 1.30, 1.31 (2t, 3JHH = 7.0 Hz, 12H, POCH2CH3); 1.50–1.63 (m, 2H, CH2); 1.74–1.91 (m, 2H, CH2P); 1.99–2.15 (m, 2H, CH2P); 2.38–2.56 (m, 1H, CHCH2P); 3.33, 3.39 (2t, 3JHH = 7.8 Hz, 2H, CH2CH2N); 3.56, 3.64 (2d, 3JHH = 7.5 Hz, 2H, CHCH2N); 4.01–4.16 (m, 8H, POCH2CH3); 5.66–5.72 (m, 1H, CH2=CH); 6.33, 6.36 (2dd, 2JHH = 2.0 Hz, 3JHH = 16.7 Hz, 1H, CH2=CH); 6.56, 6.67 (2dd, 3JHH = 10.4 Hz, 3JHH = 16.7 Hz, 1H, CH2=CH). 31P NMR (162 MHz, CDCl3): 2 rotamers δ = 29.2, 30.2. 13C NMR (101 MHz, CDCl3): 2 rotamers δ = 13.8 (2s, CH2CH2CH3); 16.4 (d, 3JCp = 6.6 Hz, POCH2CH3); 20.0, 20.3 (2s, CH2); 27.7 (dd, 1JCp = 140.3 Hz, 3JCp = 9.1 Hz, CH2P); 28.3 (t, 2JCp = 3.9 Hz, CHCH2P); 28.4 (dd, 1JCp = 139.9 Hz, 3JCp = 9.1 Hz, CH2P); 29.5 (t, 2JCp = 3.9 Hz, CHCH2P); 31.5 (s, CH2); 47.1, 47.7 (2s, CH2CH2N); 49.9, 51.9 (2t, 3JCp = 9.2 Hz, CHCH2N); 61.6, 61.7, 61.8 (4d, 2JCp = 6.6 Hz, POCH2CH3); 127.6, 127.8 (2s, CH=CH2); 128.3, 128.4 (2s, CH=CH2); 166.5, 167.0 (2s, C=O).
2.4.6.6 Synthesis of the monomers 16a and 16b
2.4.6.6.1 2-[(N-methyl)acrylamidomethyl]-1,3-propylidenediphosphonic acid 16a
The monomer 16a was prepared from the diphosphonate 15a (6.28 g, 15.2 mmol) according to the procedure described for the synthesis of the monomer 4. 4.30 g (14.3 mmol) of the desired compound were isolated.
Yield: 94%. Aspect: white solid. Mp: 130 °C.1H NMR (400 MHz, D2O): 2 rotamers δ = 1.69–1.86 (m, 2H, CH2P); 1.88–2.04 (m, 2H, CH2P); 2.36–2.55 (m, 1H, CHCH2P); 2.93, 3.08 (2s, 3H, CH3N); 3.53, 3.58 (2d, 3JHH = 7.6 Hz, 2H, CHCH2N); 5.76 (dm, 3JHH = 10.4 Hz, 1H, CH2=CH); 6.11, 6.12 (2dd, 2JHH = 1.3 Hz, 3JHH = 16.8 Hz, 1H, CH2=CH); 6.67, 6.68 (2dd, 3JHH = 10.4 Hz, 3JHH = 16.7 Hz, 1H, CH2=CH). 31P NMR (162 MHz, D2O): 2 rotamers δ = 27.5, 28.1. 13C NMR (101 MHz, D2O): 2 rotamers δ = 27.1, 28.3 (2t, 2JCp = 3.9 Hz, CHCH2P); 28.8, 29.3 (2dd, 1JCp = 135.0 Hz, 3JCp = 8.6 Hz, CH2P); 34.4, 35.7 (2s, CH3N); 52.3, 54.4 (2t, 3JCp = 10.1 Hz, CHCH2N); 127.4, 127.6 (2s, CH=CH2); 128.7, 129.0 (2s, CH=CH2); 169.6, 169.7 (2s, C=O). HRMS (m/z): calcd for C8H18NO7P2: 302.0559; found: 3025.0560 [M + H]+.
2.4.6.6.2 2-[(N-butyl)acrylamidomethyl]-1,3-propylidenediphosphonic acid 16b
The monomer 16b was prepared from the diphosphonate 15b (6.88 g, 15.1 mmol) according to the procedure described for the synthesis of the monomer 4. 5.13 g (15.0 mmol) of the desired compound were isolated.
Yield: 99%. Aspect: slightly yellow solid. 1H NMR (400 MHz, D2O): 2 rotamers δ = 0.81 (t, 3JHH = 7.4 Hz, 3H, CH2CH2CH3); 1.16–1.28 (m, 2H, CH2); 1.42–1.57 (m, 2H, CH2); 1.68–1.85 (m, 2H, CH2P); 1.86–2.04 (m, 2H, CH2P); 2.34–2.54 (m, 1H, CHCH2P); 3.33, 3.38 (2t, 3JHH = 7.7 Hz, 2H, CH2CH2N); 3.52, 3.56 (2d, 3JHH = 7.5 Hz, 2H, CHCH2N); 5.73 (dm, 3JHH = 10.6 Hz, 1H, CH2=CH); 6.11 (dm, 3JHH = 16.9 Hz, 1H, CH2=CH); 6.67, 6.68 (2dd, 3JHH = 10.4 Hz, 3JHH = 16.7 Hz, 1H, CH2=CH). 31P NMR (162 MHz, D2O): 2 rotamers δ = 27.6, 28.2. 13C NMR (101 MHz, D2O): 2 rotamers δ = 12.9, 13.0 (2s, CH2CH2CH3); 19.1, 19.4 (2s, CH2); 27.5, 28.5 (2t, 2JCp = 3.7 Hz, CHCH2P); 28.7, 29.2 (2dd, 1JCp = 135.7 Hz, 3JCp = 9.1 Hz, CH2P); 30.4 (s, CH2); 46.7, 47.9 (2s, CH2CH2N); 50.2, 52.0 (2t, 3JCp = 9.8 Hz, CHCH2N); 127.4, 127.5 (2s, CH=CH2); 128.9, 129.0 (2s, CH=CH2); 169.0, 169.3 (2s, C=O). HRMS (m/z): calcd for C11H24NO7P2: 344.1028; found: 344.1043 [M + H]+.
2.5 Photopolymerization procedure
Photopolymerizations were carried out on a Perkin–Elmer differential scanning calorimeter (DSC), Pyris Diamond. 0.5 wt.% of BMDG (photoinitiator) were added to each comonomer mixture. A sample (ca. 0.8 mg) of each mixture was placed in an uncovered aluminum DSC pan. The DSC chamber was purged with nitrogen for 5 min before polymerization. One minute after the beginning of the acquisition, the samples were irradiated for 2 min at 37 °C with a LED curing light (Bluephase, Ivoclar-Vivadent AG). The incident light intensity was 20 mW/cm2. Each experiment was repeated three times. The heat flux was monitored as a function of time using the DSC under isothermal conditions. The double-bond conversion (DBC) was calculated as the quotient of the overall enthalpy evolved [ΔHP (J/g)] and the theoretical enthalpy obtained for 100% conversion of the mixtures [ΔH0P (J/g)] (Equation (1)).(1)
ΔH0P was calculated according to the following formula (Equation (2)):(2) where ΔH0i is the theoretical enthalpy of monomer i (i = methacrylate, ΔH0i = 54.8 kJ/mol Citation[17]; i = monoacrylamide, ΔH0i = 60.3 kJ/mol; i = diacrylamide, ΔH0i = 120.6 kJ/mol Citation[18]), Mi its molar mass and Pi the amount used in the formulation (wt.%).
The rate of polymerization (Rp) was calculated according to the following formula (Equation (3)):(3) where Q is the heat flow per second during the reaction and m the mass of the mixture in the sample.
2.6 Shear bond strength measurement
Freshly extracted bovine mandibular incisors were embedded in unsaturated polyester resin (Castolite). Flat dentinal and enamel surfaces were prepared with 120-grit and 1000-grit, wet silicon carbide paper on the labial side of the embedded teeth. The primer was first rubbed on the prepared surface (dentin or enamel) with a microbrush for 15 s and let undisturbed for 15 s. The primer layer was strongly air dried. The AdheSE bonding agent was subsequently applied with a microbrush. The adhesive was light cured for 10 s with a LED curing light (Bluephase G20, Ivoclar Vivadent AG). A 3 mm thick cylindrical Teflon mold with a central 2 mm diameter circular hole was fixed on the surface. A composite (Tetric EvoCeram, Ivoclar Vivadent AG) was inserted in the mold and light-cured for 20 s. The samples were finally stored in water at 37 °C for 24 h before being tested. The shear bond strength was measured using a universal testing machine (Zwick) at a crosshead speed of 0.8 mm/min. 10 samples were tested for each adhesive.
3 Results and discussion
3.1 Syntheses
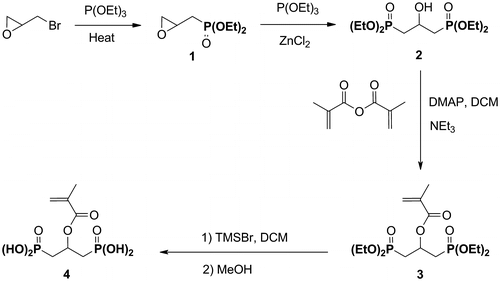
The acidic monomer 4 was prepared in four steps, starting from epibromohydrine (Scheme ). The diethyl 2,3-epoxypropylphosphonate 1 was first obtained by a Michaelis–Arbuzov reaction, according to a procedure reported in the literature.Citation[14] The diphosphonate 2 was subsequently synthesized by a regioselective opening of 1 with TEP in the presence of ZnCl2.Citation[19] It was isolated in 48% yield. The diphosphonate 2 was then reacted with methacrylic anhydride in the presence of TEA and of a catalytic amount of DMAP. This reaction was carried out under reflux conditions. After purification by flash column chromatography, the desired monomer was isolated in 85% yield. Finally, the diphosphonate 3 was silylated using an excess of TMSBr (6.0 eq.). The methanolysis of the silyl ester provided the expected acidic monomer 4 in 97% yield.
Scheme 1 Synthesis of the acidic monomer 4.
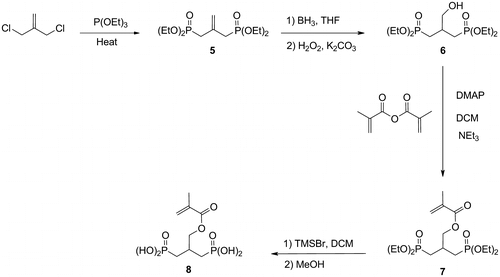
The diphosphonic acid 8 was synthesized in four steps (Scheme ). Tetraethyl 2-methylene-1,3-propylendiphosphonate 5 was first prepared by a Michaelis-Arbusov reaction.Citation[15] The diphosphonate 6 was then prepared according to the conditions reported by Cattaneo et al. Citation[16]. The hydroboration of 5 using a borane-tetrahydrofuran complex, followed by an oxidation involving hydrogen peroxide, afforded 6 in good yield. An acylation, followed by the deprotection of the phosphonate groups, finally led to the desired diphosphonic acid 8.
Scheme 2 Synthesis of the acidic monomer 8.
The acidic monomers 10a and 10b were synthesized in two steps, starting from 6 (Scheme ). The carbamate 9a was easily obtained by reacting 6 with IEM in the presence of a catalytic amount of dibutyltin dilaurate. The monomer 9a was isolated, after purification, in a 86% yield. The synthesis of 9b required in the first place the preparation of the 10-isocyanatodecyl methacrylate. This isocyanate was prepared by the condensation of potassium cyanate with 10-bromodecyl methacrylate in MeCN, using NBu4Br as a phase transfer catalyst.Citation[20] Although 10-isocyanatodecyl methacrylate was mainly obtained, secondary reactions also led to the formation of both the corresponding isocyanurate, coming from the trimerization of the isocyanate, as well as to the corresponding urea derivative. Afer work-up, the resulting crude product was directly reacted with 6 in order to obtain 9b in good yield. The deprotection of the phosphonate groups of 9a and 9b, respectively, led to 10a and 10b in excellent yields.
Scheme 3 Synthesis of the acidic monomers 10a and 10b.
It is well known that methacrylates are unstable under aqueous acidic conditions.[21,22]Citation21Citation22 Indeed, a hydrolysis of the ester group takes slowly place. In order to improve the stability of SEAs, monomers exhibiting a better stability towards hydrolysis were synthesized. Hence, monomers in which the polymerizable acrylate group is connected to the phosphonic acid group via a hydrolytically stable ether bond were prepared.[23,24]Citation23Citation24 It was also demonstrated that N-alkylacrylamides exhibit a great stability in such acidic conditions.[2,9,10,25]Citation2Citation9Citation10Citation25 Monomers 4, 8, 10a and 10b bearing a methacrylate group, their storage in aqueous solutions would require low temperatures (storage in the refrigerator). In this context, we took an interest in the synthesis of the monomers 12, 16a and 16b, which exhibit a higher stability in aqueous media (Figure ).
The diphosphonic acid 12 was synthesized from 6 in 2 steps (Scheme ). The monomer 11 was first obtained by the reaction of 6 with ethyl 2-(chloromethyl)-acrylate, in the presence of TEA. The reaction was carried out in THF, for 45 h, at 70 °C. After purification, 11 was isolated in moderate yield (48%). The deprotection of the phosphonate groups afforded 12 in a quantitative yield.
Scheme 4 Synthesis of the acidic monomer 12.
Finally, the monomers 16a and 16b were prepared in 4 steps from the diphosphonate 6 (Scheme ). The hydroxyphosphonate 6 was reacted with methanesulfonyl chloride, in the presence of TEA, to afford 13 in 85% yield. A nucleophilic substitution using a large excess of the corresponding amine (methylamine or butylamine) was then carried out in ethanol. The aminobisphosphonates 14a and 14b were isolated in respectively 91% and 84% yields. An acylation using acryloyl chloride, followed by a deprotection of the phosphonate groups, led to the expected diphosphonic acids 16a and 16b in good yields.
Scheme 5 Synthesis of the acidic monomers 16a and 16b.
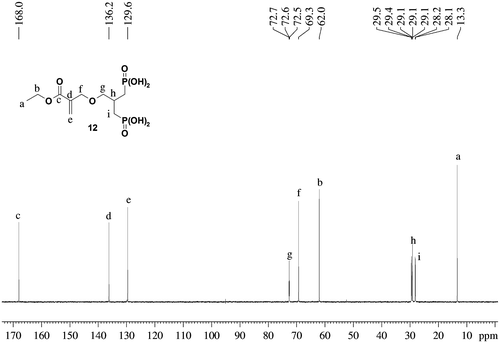
The structures of the new acidic monomers were confirmed by 1H NMR, 31P NMR, 13C NMR spectroscopy as well as by HRMS. As an example, the 13C NMR spectrum of monomer 12 is represented on Figure . The monomer 12 is identified by four methylene groups at 28.8 (2C, dd), 62.0, 69.3 and 72.6 ppm (t) as well as by a methyl group at 13.3 ppm and a carbonyl carbon at 168.0 ppm. The carbon in β position of the phosphonic acid groups is characterized by a triplet at 29.1 ppm. The carbons of the acrylate double bond are identified by the presence of two singlets at 129.6 and 136.2 ppm.
Figure 3 13C NMR spectrum of monomer 12.
3.2 Photopolymerizations
To investigate their reactivities, each synthesized acidic monomer was copolymerized with DEBAAP. Most of the diphosphonic acids being not soluble in DEBAAP, DMF was added as a solvent. Formulations containing 30 mol% of DEBAAP, 20 mol% of an acidic monomer and 50 mol% of DMF were prepared. 0.5 wt.% of BMDG were introduced as a photoinitiator. It has been recently demonstrated that this germanium initiator exhibits a high photoinitiation activity as well as an outstanding photobleaching activity.Citation[26] All photopolymerizations were performed under the same conditions (irradiation time: 2 min; light intensity: 20 mW/cm2). Results are reported in Table . For each copolymerization, the maximal rate of polymerization (Rpmax), the time of irradiation needed to reach this maximum () and the double bond conversion (DBC) are indicated. These results showed that each acidic monomer was able to efficiently copolymerize with DEBAAP. Although monomers 4, 8, 10a, 10b, 16a and 16b exhibited a similar reactivity in copolymerization with DEBAAP, monomer 12 was somewhat less reactive. It might be explained on the basis of steric effects.
Table 1. Rpmax, and DBC measured for different comonomer mixtures.
3.3 Adhesive properties
The adhesive properties of the synthesized monomers were investigated. These diphosphonic acids were evaluated in 2-step self-etching adhesives. Self-etching primers 4, 8, 10a, 10b, 16a and 16b, containing the corresponding acidic monomer as well as a cross-linking co-monomer (DEBAAP), water, ethanol, a photoinitiator (CQ) and a stabilizer (BHT), were formulated (Table ). A primer based on 1,3-bis(methacrylamido)propane-2-yl dihydrogen phosphate 17 was also prepared. This monomer can be found in some commercially available adhesives (AdheSE One, Ivoclar Vivadent AG, Liechtenstein). Therefore, this primer was used as a reference. Each primer was coupled with the AdheSE bonding agent in order to achieve a bond between the dental hard tissues (dentin and enamel) and a restorative material (Tetric EvoCeram). The bonding agent is a hydrophobic resin which is mainly made of a mixture of dimethacrylates and HEMA. The results of the dentin and enamel shear bond strength tests are reported in Table . Except for compound 16a, all monomers were able to generate a strong bond between the dental tissues and the composite. Surprisingly, the application of the primer 16a led to a premature rupture of the bond. Similar results were reported in the literature when using 3-(N-propylacrylamido)propylidenebisphosphonic acid as an acidic monomer.Citation[10] This result was attributed to an incompatibility between the primer and the bonding resin. Primers 4, 8, 10a and 10b led to similar dentin SBS than the formulation 17. However, primers 8, 10a and 10b conducted to significantly higher enamel SBS than 17. Monomers 10a and 10b showed the best results. These observations could partially be attributed to a better etching ability of the corresponding monomers, due to the presence of two phosphonic acid groups. Besides, the additional hydrogen bonding interactions due to the presence of the carbamate group might improve the quality of the adhesion. Unfortunately, the use of the hydrolytically stable monomers 12 and 16b resulted in significantly lower dentin SBS. Moreover, 16b was significantly less efficient than the other synthesized monomers regarding enamel adhesion. From this study, it can be asserted that methacrylates 4, 8, 10a and 10b have the potential to be used in new dental formulations.
Table 2. Composition of the primer.
Table 3. Shear bond strength of the different adhesive systems to dentin and enamel.
4 Conclusion
A series of polymerizable 2-substituted-1,3-propylidenediphosphonic acids were synthesized for an application in dental materials. The photopolymerization behavior of these monomers with DEBAAP in DMF was investigated by photo-DSC. All monomers were able to efficiently copolymerize with DEBAAP. Monomers 4, 8, 10a, 10b, 16a and 16b exhibited no significant difference of reactivity in copolymerization with DEBAAP. However, the mixture containing the monomer 12 was significantly less reactive. New SEAs based on these diphosphonic acids were formulated. Apart from the formulation based on the monomer 16a, which did not enable the formation of a bond between the dental tissues and the dental restoration, all adhesives led to high dentin and enamel SBS. Monomers 8, 10a and 10b leading to similar dentin SBS as well as to significantly higher enamel SBS than monomer 17, which is incorporated in some commercially available formulations, are therefore great candidates for the formulation of new dental materials.
Notes
1Part 12: cf. Ref. Citation[1].
Related Research Data
References
- Pavlinec J, Kleinova A, Angermann J, Lamparth I, Moszner, N. Monomers for adhesive polymers, 12. Synthesis and free-radical homo- and copolymerization of 2-ethoxycarbonyl-allyl 5-(1,2-dithiolane-3-yl)-pentanoate. Macromol. Mater. Eng. doi:10.1002/mame.201200382.
- Moszner N, Hirt T. New polymer-chemical developments in clinical dental polymer materials: enamel–dentin adhesives and restorative composites. J. Polym. Sci., Part A: Polym. Chem. 2012;50:4369–4402.
- Van Meerbeek B, Yoshihara K, Yoshida Y, Mine A, De Munck J, Van Landuyt KL. State of the art of self-etch adhesives. Dental Mater. 2011;27:17–28.
- Van Landuyt KL, Snauwaert J, De Munck J, Peumans M, Yoshida Y, Poitevin A, Coutinho E, Suzuki K, Lambrechts P, Van Meerbeek B. Systematic review of the chemical composition of contemporary dental adhesives. Biomaterials. 2007;28:3757–3785.
- Van Landuyt KL, Yoshida Y, Hirata I, Snauwaert J, De Munck J, Okazaki M, Suzuki K, Lambrechts P, Van Meerbeek B. Influence of the chemical structure of functional monomers on their adhesive performance. J. Dental Res. 2008;87:757–761.
- Yoshida Y, Van Meerbeek B, Nakayama Y, Yoshioka M, Snauwaert J, Abe Y, Lambrechts P, Vanherle G, Okazaki M. Adhesion to and decalcification of hydroxyapatite by carboxylic acids. J. Dental Res. 2001;80:1565–1569.
- Yoshioka M, Yoshida Y, Inoue S, Lambrechts P, Vanherle G, Nomura Y, Okazaki M, Shintani H, Van Meerbeek B. Adhesion/decalcification mechanisms of acid interactions with human hard tissues. J. Biomed. Mater. Res. 2002;59:56–62.
- Catel Y, Fischer U, Moszner N. Monomers for adhesive polymers, 10. Synthesis, radical photopolymerization and adhesive properties of methacrylates bearing phosphonic acid groups. Macromol. Mater. Eng. 2013;298:740–756.
- Moszner N, Angermann J, Fischer U, Bock T. Monomers for adhesive polymers, 9. Synthesis, radical photopolymerization, and properties of (meth)acrylamido dihydrogen phosphates. Macromol. Mater. Eng. 2013;298:454-461.
- Catel Y, Degrange M, Le Pluart L, Madec PJ, Pham TN, Chen F, Cook WD. Synthesis, photopolymerization, and adhesive properties of new bisphosphonic acid monomers for dental application. J. Polym. Sci., Part A: Polym. Chem. 2009;47:5258–5271.
- Catel Y, Besse V, Zulauf A, Marchat D, Pfund E, Pham TN, Bernache-Assolant D, Degrange M, Lequeux T, Madec PJ, Le Pluart L. Synthesis and evaluation of new phosphonic, bisphosphonic and difluoromethylphosphonic acid monomers for dental application. Eur. Polym. J. 2012;48:318–330.
- Akgun B, Savci E, Avci D. Synthesis and polymerizations of phosphonated-bis(methacrylamide)s for dental applications. J. Polym. Sci., Part A: Polym. Chem. 2012;50:801–810.
- Ikemura K, Kadoma Y, Endo T. A review of the developments of self-etching primers and adhesives – Effects of acidic adhesive monomers and polymerization initiators on bonding to ground, smear layer-covered teeth. Dent. Mat. J. 2001;30:769–789.
- Phillips AMMM, Mphahlele ML, Modro AM, Modro TA, Zwiezak A. Phosphonic systems, 7. Reactions of 2,3-epoxyphosphonates with nucleophiles: preparation of 2,3-disubstituted alkylphosphonic esters and related systems. Phosphorus, Sulfur Silicon Relat. Elem. 1992;71:165–174.
- Bodesheim F, Velker E, Bentz F, Nischk G. Reaktionen von 2-methylen-propan-1,3-diol-derivaten mit organischen phosphorverbindungen. Chemiker-Zeitung. 1972;96:581–582.
- Cattaneo M, Lecchi A, Ohno M, Joshi BV, Besada P, Tchilibon S, Lombardi R, Bischofberger N, Harden TK, Jacobson KA. Antiaggregatory activity in human platelets of potent antagonists of the P2Y1 receptor. Biochem. Pharmacol. 2004;68:1995–2002.
- Anseth KS, Wang CM, Bowman CN. Kinetic evidence of reaction-diffusion during the polymerization of multi(meth)acrylate monomers. Macromolecules. 1994;27:650–655.
- Ulrich G, Burtscher P, Salz U, Moszner N, Liska R. Phenylglycine derivatives as coinitiators for the radical photopolymerization of acidic aqueous formulations. J. Polym. Sci., Part A: Polym. Chem. 2006;44:115–125.
- Sardarian AR, Shahsavari-Fard Z. Convenient and regioselective one-pot solvent-free synthesis of ß-hydroxyphosphonates. Synth. Commun. 2007;37:289–295.
- Dubosclard-Gottardi C, Caubère P, Fort Y. New selective syntheses of (meth)acrylic monomers: isocyanates, isocyanurates, carbamates and ureas derivatives. Tetrahedron. 1995;51:2561–2572.
- Salz U, Zimmermann J, Zeuner F, Moszner N. Hydrolytic stability of self-etching adhesive systems. J. Adhes. Dent. 2005;7:107–116.
- Teshima I. Degradation of 10-methacryloyloxydecyl dihydrogen phosphate. J. Dental Res. 2010;89:1281–1286.
- Moszner N, Zeuner F, Pfeiffer S, Schurte I, Rheinberger V, Drache M. Monomers for adhesive polymers, 3. Synthesis, radical polymerization and adhesive properties of hydrolytically stable phosphonic acid monomers. Macromol. Mater. Eng. 2001;286:225–231.
- Klee JE, Lehmann U. Novel 2-(ω-phosphonooxy-2-oxaalkyl)acrylate monomers for self-etching self-priming one part adhesive. Beilstein J. Org. Chem. 2010;6:766–772.
- Catel Y, Degrange M, Le Pluart L, Madec PJ, Pham TN, Picton L. Synthesis, photopolymerization and adhesive properties of new hydrolytically stable phosphonic acids for dental applications. J. Polym. Sci., Part A: Polym. Chem. 2008;46:7074–7090.
- Moszner N, Zeuner F, Lamparth I, Fischer U. Benzoylgermanium derivatives as novel visible-Light photoinitiators for dental composites. Macromol. Mater. Eng. 2009;12:877–886.