
Abstract
The solution and micellar polymerizations of acrylamide (Aam) initiated by the photoinitiator DAROCUR 2959 (DAR) under UV-light were studied. Variations of kinetic parameters with DAR and AAm concentrations and with the emulsifier type were followed. Furthermore, the kinetics of AAm polymerization under argon and air atmosphere was investigated. Dilatometry was implemented to follow continuously the fast photopolymerization. The polymerization rate vs. conversion curve of the photoinduced polymerization of AAm was described by two and four nonstationary rate intervals. Variation of the rate of polymerization with conversion was discussed in terms of two types of initiating radicals and the gel effect. They are formed by the decomposition of excited DAR* {DAR…AAm}* or {DAR…O2}* exciplexes. The intra- and intermolecular association of polyAAm, the self-assembling of AAm monomers, and the association of PAAm with Aam and cross-linking change the solution polymerization into the dispersion one.
Experimental
Materials
Commercially available acrylamide (AAm, Fluka) was used as supplied. Extra pure Darocur 2959 (DAR) (2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone, C12H16O4, Mw = 224.25, Figure ) was used as supplied by Fluka. Nonionic emulsifiers used were the reagent-grade Tween 60 (polyoxyethylene sorbitan oleate, provided by Serva, Tw60, hydrophilic–lipophilic balance (HLB) = 14.9) and Tween 85 (polyoxyethylene sorbitan trioleate, provided by Serva, Tw85, HLB = 11.0). Doubly distilled water used as a reaction medium was deprived of oxygen by heating to boiling point and cooling under a stream of argon.
The n–π* transitions for DAR in water are expected at 331–365 nm.[Citation1] DAR being a liquid can be expected to impart a good diffusion of reactive radical species and hence a high conversion can be obtained for the formulation with DAR (its molar extinction coefficient at 365 nm in water was estimated to be ca. 10.0 dm3/mol.cm).
Polymerization procedure
The polymerization experiments were performed on an optical bench using UV-light of wavelengths λ = 365 nm with intensity Io = 3.5 × 10−6 einstein dm−3 s−1 at 23 °C. Amounts of reactants (DAR, AAm, Tw60, and Tw85) were used as shown later. The polymerization technique, conversion determination (dilatometric and gravimetric techniques), the estimation of polymerization rate, and the measurements of light intensity were the same as described earlier.[Citation2–Citation4] Some details of these techniques are summarized below.
The preparation of the solutions for dilatometric measurements comprised weighing the respective reactants, putting it into a volumetric flask, and filling this flask with water up to the mark. The solutions thus prepared were bubbled through with argon and immediately used for polymerization.
The dilatometer was cylindrical in shape with parallel walls of quartz glass, and had an internal diameter of 2.70 cm and a thickness of 1.5 cm. It contained a capillary with 0.123 cm inside diameter, and the total irradiated volume was 7.84 ml. In some instances, cells of 0.4 cm path length equipped with a capillary of 0.5 mm inside diameter were used. Argon was bubbled through the dilatometer before use, and the reaction mixture was introduced into the dilatometer through a capillary by means of a syringe. After filling the dilatometer with polymerization solution, a three-way cock was joined with the ground glass capillary terminal and connected with water pump and argon reservoir. After cooling the dilatometer with a solid CO2–methanol mixture, the vacuum water pump was applied by opening a stop-cock momentarily. Another turn of the stopcock canceled the evacuation by letting in argon. The dilatometer was gradually heated to room temperature and the whole process involving cooling and evacuation was repeated three times. This procedure ensured the removal of bubbles in the dilatometer as well as the inert medium (argon) from the polymerization solution. After the last cycle was finished, the dilatometer with the stopcock closed was put on the optical bench and thermostatted to the temperature of polymerization prior to irradiation. The dilatometer was placed on an optical bench in a temperature-controlled metal block.
The movement of the meniscus in the capillary of the dilatometer was followed by means of a cathetometer. The conversion was calculated from the relation x = (Vo − Vt)/Vo × K, where Vo and Vt represent the total volume of the monomer at time zero and time t of polymerization and K is the shrinkage constant of monomer. The conversion (x) – time (t) data were graphically differentiated to yield the rate of polymerization Rp = (dx/dt) as a function of conversion. The value of the conversion was calculated from the relation x = 1 – nt/no, where no and nt represent the total amout of monomer (in mol) in the feed at time zero and time t of polymerization. The conversion determined dillatometrically was correlated with that determined gravimetrically (x (%) = mpol/mmon × 100, where mpol/represents the amount of polymer formed at time t and mmon represent the initial amount of monomer).
A spherical high-pressure mercury lamp DRS 1000 (Institute of Chemical Physics, Moscow, USSR) was used as a radiation source. It emitted rays in the ultraviolet and the visible region with the wavelengths 289.4 nm (5.8), 296.7 nm (11.7), 302.2 nm (17.8), 312.6 nm (28.3), 313.2 nm (28.9), 334 nm (34.7), 365 nm (84.4), 365.5 nm (75.2), 404.7 nm (72.8), 407.8 nm (23.3), 435.8 nm (100), 546.1 nm (69.4), 577 nm (54.4), 579 nm (47.6). The values in parentheses give the intensity of individual lines relative to the intensity I at 435.8 nm. In order to select the wavelength of 365 nm, a combination of liquid and glass filters was used. The filters were placed in the cooling jacket of the optical bench. A beam of parallel rays impinging on the dilatometer passed through a diaphragm of 2.58 cm diameter.
The intensity of incident rays Io was measured by means of a quanta counter. The fluorometric cell of the quata counter was filled with an ethylene glycol solution of Rhodamine B. Two photodiodes connected in series were placed on both sides of the cell in the proximity of the front window of the cell. The direct current from photodiodes was measured by microgalvanometer. The microgalvanometer reading was calibrated with a ferrioxalate actinometer and checked by radiometer. If needed, the intensity of the rays entering the dilatometer was adjusted by means of metal gauze and glass filters with different transmissions and partly regulating the voltage in discharge tube. The glass filters were placed in the cooling jacket of the optical bench.
Introduction
Water-soluble polymers have received the considerable interest of scientists and engineers working on environmental and industrial problem. Polyacrylamide and its copolymers comprise the majority of the market, which includes applications in paper making, the flocculation of municipal and industrial waste water, mineral processing, water treatment, metal salt reduction, particle formation and its stabilization, and as various composite adhesives. AAm monomers are completely soluble in water. The properties of final polymers and copolymers are regulated by a broad choice of additives. A vast majority of the studies on solution and micellar polymerization (McP) processes focus primarily on thermally initiated polymerization.
The polymer/micellar association is well known [Citation5] in the literature. The thermally initiated polymerization can be effected by means of electrostatic and/or hydrophobic interactions. For example, the addition of anionic emulsifiers (above its CMC) was found to increase the rate of polymerization of AAm.[Citation6,Citation7] The nonionic emulsifier did not effect the polymerization rate of AAm. A cationic emulsifier caused even a decrease in the rate of polymerization. In the presence of anionic emulsifier (Dowfax 2A1), the rate of polymerization of AAm was independent of Dowfax 2A1.[Citation8] The anionic emulsifier worked only as an efficient chain transfer agent. In the presence of poly(butyl acrylate) particles, however, an increase of the polymerization rate was observed. That was suggested to be consequence of the grafting to the polymer particles with AAm. On the contrary, the rate of polymerization increased and the degree of polymerization decreased in the presence of emulsifiers C8H17SO3Na, C12H25SO3Na, and C16H25SO3Na.[Citation9]
A first use of the photoinduced polymerization was mentioned in 1989 by Candau et al. [Citation10]. Somewhat later I. Capek investigated the photoinduced emulsion (co)polymerization of alkyl(acrylates).[Citation11,Citation12] The more-detailed photoinduced miniemulsion polymerization was mentioned in 2009 by Tonar et al. [Citation13] who investigated the controlled radical polymerization of vinyl acetate in the presence of an iodinated macrophotoinitiator. Despite incomplete conversion and long polymerization times, the controlled character of the reaction was demonstrated. Chemtob et al. have presented an interesting approach which is very different from those in above since conventional free radical photopolymerization in micellar systems was investigated with a formulation including simply conventional acrylate monomers (butyl acrylate, methyl methacrylate, acrylic acid) and a commercial type of radical photoinitiatiors.[Citation14] A range of commercial water-soluble and water-insoluble radical photoinitiators proved to be well suited to initiate the polymerization of a nanosized acrylate miniemulsion in the presence of a conventional anionic surfactant.
The synthesis of hydrophobically modified polyacrylamides by means of a McP process was described.[Citation15] In aqueous solution, above a certain concentration, the hydrophobic groups associate intermolecularly and build up a transitory network. In this process, the hydrophobic monomer is solubilized within the surfactant micelles, whereas the hydrophilic monomer is dissolved in the aqueous continuous phase. This method of synthesis leads to water-soluble polymers containing randomly distributed hydrophobic blocks along the hydrophilic backbone. This behavior prompted us to study the effect of hydrophilic and hydrophobic surfactant molecules on the photoinduced radical polymerization of AAm in the micellar solution. We have used both the hydrophobic Tween 85 surfactant based on polyethyleneoxide (PEO) with trioleoyl lipid group and the hydrophilic Tween 60 surfactant based on PEO with oleoyl lipid group. The aim of the present study is to find out the influence of the micelles of hydrophobic and hydrophilic structurally similar emulsifiers (Tween 85 and Tween 60) on the aqueous phase polymerization of AAm photoinitiated by DAR photoinitiator. The presence of emulsifier micelles models the polymerization process under partitioning of precursors and reactants between aqueous and micellar phases. Besides, the emulsifier molecules and their associates are expected to influence the intermolecular association of polyAAm chains during the polymerization.
Under the ideal solution polymerization, the long stationary-state rate interval is supposed to appear. When the polymerization proceeds under the self-association of monomers and their polymers and/or the interaction between monomer with its polymer (a template polymerization), then the deviation from the ideal polymerization can be observed. Under such conditions, the dependence of rate of polymerization on conversion might be similar to that observed in a dispersion system. In our previous works, we observed that the dependence of the polymerization rate vs. conversion in the some polymerization systems is desribed by the two or even by four rate intervals with one or two rate maxima.[Citation16,Citation17] The aim of this study is to model the AAm polymerizations with several rate intervals. We expect that the single radicals derived from the DAR start the polymerization and the formed polyAAm associates itself and forms also aggregates with its monomer. The first rate maximum appear as a result of polymerization within the monomer/polymer aggregates. The second rate maximum should appear by the postponed formation of radicals. The effect of gel effect on the appearance of the second maximum is not excluded. It is expected that during the photopolymerization under oxygen, the second type of radicals (peroxides) accumulate.
Results and discussion
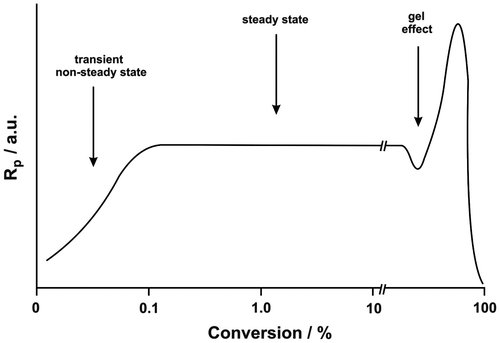
The rate of polymerization vs. conversion curve typical for the homogeneous polymerization consists of several parts; one linear part up to medium or high conversion (stationary-state interval), the second one governed by the gel effect (concave upward) and the last concave downward (the diffusion-controlled step) (Figure ).[Citation18] At the beginning, there is a short period when the rate is changing and the reaction reaches a steady state; as the reaction advances to medium or high conversion, both the rate and the degree of polymerization are observed to pass through minima. This is regarded as the onset of the “gel effect;” a period when the increasing viscosity of the reaction system causes the reaction to accelerate as well. The rate of acceleration is matched by an increase in the molecular weigh and its distribution. Ultimately, the rate of polymerization passes through a maximum; as the mixture becomes glassy, the rate falls to nearly zero at some conversion less than 100%.[Citation19] Kinetic model of free radical polymerization involves the unimolecular decomposition of initiator I, to produce two free radicals, R, a fraction of which, f, then add monomer, M, to initiate the active polymer chain, P. Active chain either propagates and grows by adding more monomer or stops its growth by undergoing chain transfer with some species Y or by termination in a bimolecular reaction with another active chain.
Figure 1. Structure of DAROCUR 2959.
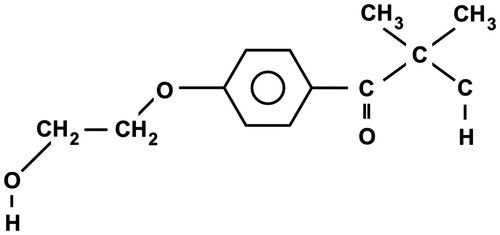
Figure 2. Rate of ideal solution radical polymerization as a function of conversion.
The photoinduced micellar and solution radical polymerizations of AAm under air or inert (argon) conditions are summarized in Figures –. The shape of all conversion curves deviates from the shape typical for the ideal solution polymerizations. Its shape is more typical for the precipitation or dead end polymerization. The conversion curves are concave downward nearly throughout the polymerization (Figures and ). The linear part of conversion curve for the aqueous polymerization of AAm is relatively short or it does not appear at all.
Figure 6. Variation of the rate of photoinduced solution polymerization of AAm with conversion and bubbling time. Recipe: 1.5 g AAm, 27 g H2O, 0.0515 g DAR, 1) bubbling time = 0 min, 2) bubbling time = 3 min.
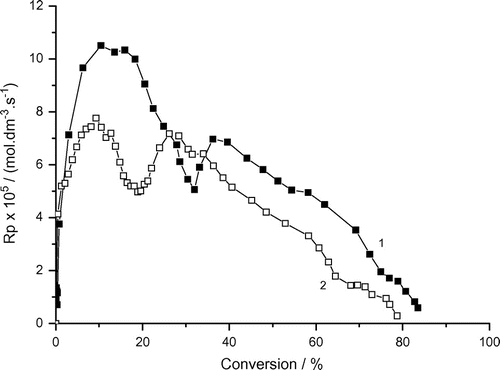
Figure 5. Variation of the rate of photoinduced micellar polymerization of AAm with conversion and bubbling time. Recipe: 1.5 g AAm, 27 g H2O, 0.36 g Tw60, 0.1030 g DAR, 1) under air, 2) under argon.
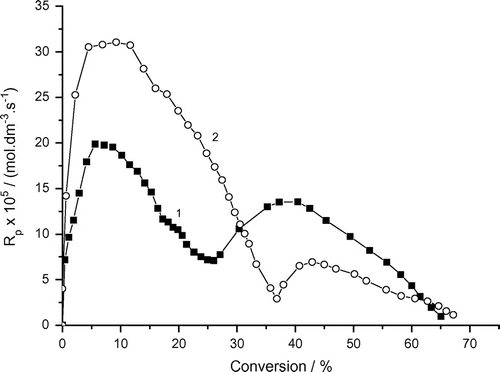
Figure 4. Variation of monomer conversion of photoinduced solution polymerization of AAm with reaction time and bubbling time. Recipe: 1.5 g AAm, 27 g H2O, 0.0515 g DAR, 1) bubbling time = 0 min, 2) bubbling time = 3 min.
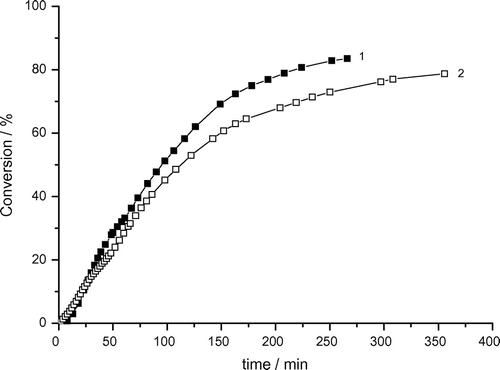
Figure 3. Variation of monomer conversion of photoinduced micellar polymerization of AAm with reaction time and bubbling time. Recipe: 1.5 g AAm, 27 g H2O, 0.36 g Tw60, 0.1030 g DAR, 1) bubbling time = 0 min, 2) bubbling time = 3 min.
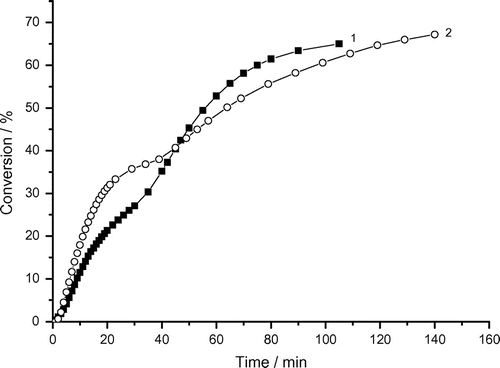
Variations of the rates of micellar radical polymerization of AAm with conversion under air (with oxygen) or argon (without oxygen) are demonstrated in Figure (data taken from Figure ). The data indicate that this dependence is described by the four rate intervals with two rate maxima (Rp, max1 (under argon (1)) and Rp, max2 (under air (2)) (Figure ). These data show that the Rp, max1 decreases and Rp, max2 increases under air (with oxygen) and the rate maxima are slightly shifted to lower conversion:
Rp, max1 = 3.1 × 10−4 mol dm−3 s−1 at 9.2% conversion, Rp, max2 = 0.75 × 10−4 mol dm−3 s−1 at 42% conversion (under argon).
Rp, max1 = 2.0 × 10−4 mol dm−3 s−1 at 5.6% conversion, Rp, max2 = 1.4 × 10−4 mol dm−3 s−1 at 38% conversion (under air).
The inhibition or retardation effect of oxygen is generally known. In our case, the rate of polymerization (Rp, max1) decreased from 3.1 × 10−4 mol dm−3 s−1 to 2.0 × 10−4 mol dm−3 s−1 but the second rate maximum (Rp, max2) increased from 0.75 × 10−4 mol dm−3 s−1 to 1.4 × 10−4 mol dm−3 s−1.
The linear part of the rate of polymerization vs. conversion curve (the stationary-state interval) was not observed in the solution radical polymerization of AAm (without emulsifier). On the contrary, the four rate intervals were found which are typical for the McP systems (Figure ):
Rp, max1 = 0.8 × 10−4 mol dm−3 s−1 at 9.3% conversion, Rp, max2 = 0.7 × 10−4 mol dm−3 s−1 at 25% conversion (under argon).
Rp, max1 = 1.05 × 10−4 mol dm−3 s−1 at 10.4% conversion, Rp, max2 = 0.7 × 10−4 mol dm−3 s−1 at 35% conversion (under air).
Furthermore, the first maximum rate Rp, max1 increased and the second one Rp, max2 did not change under air. The unexpected behavior might be discussed in terms of the formation of {DAR…oxygen}* complex and its decomposition to the initiating radicals. The consumption of oxygen at higher conversion leads to the unchanged Rp, max2. Comparng the Rp, maxs in Figures and show that the McP is faster than the solution polymerization. This indicates that the monomer swollen micelles contribute to the whole polymerization rate with the depressed termination and/or the increased monomer concentration at the reaction loci.
The effect of the photoinitiator DAR on the solution radical polymerization of AAm is summarized in Figures and . The conversion curves are concave downward and the polymerization enters to the limiting conversion at ca. 80% conversions or at ca. 250 min. The limiting conversion cannot be ascribed to the consumption of initiator because the photobleaching of DAR is negligible even after several hours. The appearance of the limiting conversion can be discussed in terms of the low monomer concentration at the reaction loci and the self-quenching of excited states of DAR including their nonradiative decay. The shape of conversion curves deviates from the shape typical for the solution polymerization of acrylate monomers. Its shape is more typical for the micellar or precipitation polymerization.
Figure 8. Variation of monomer conversion of photoinduced polymerization of AAm with reaction time and AAm concentrations. Recipe: 27 g H2O, 0.504 g Tw85, 0.103 g DAR 1) 0.5 g AAm, 2) 1.0 g AAm , 3) 1.5 g AAm, 4) 2.0 g AAm.
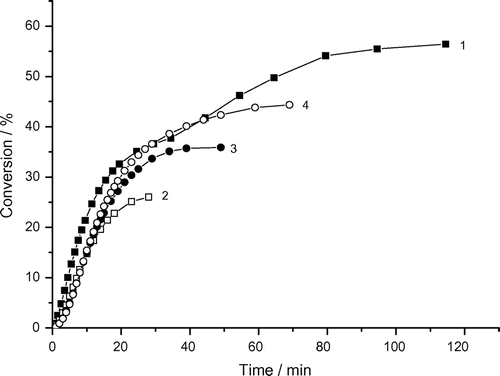
Figure 7. Variation of monomer conversion of photoinduced polymerization of AAm with reaction time and DAR concentrations. Recipe: 1.5 g AAm, 27 g H2O, 1) 0.01285 g DAR, 2) 0.02575 g DAR, 3) 0.0515 g DAR, 4) 0.1030 g DAR, 5) 0.2060 g DAR (under argon).
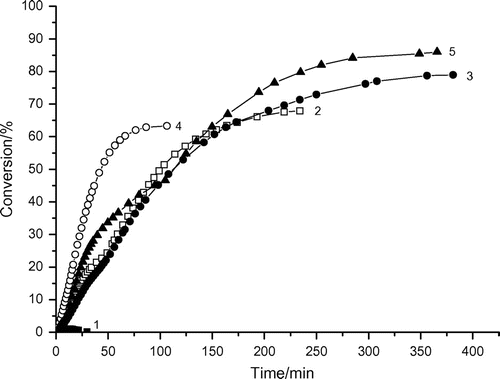
The dependence of the rate of polymerization vs. conversion listed in Figure is described by a curve with the four or two rate maxima. This behavior strongly deviates from the ideal thermally initiated solution polymerization of acrylic ester monomers which consists of one stationary-state interval and several nonstationary intervals (Figure ). The polymer/AAm aggregates start to compete for radicals with AAm from ca 5–10% conversion where the deviation from linearity appears and the polyAAm accumulates in the reaction system. The accumulation of polymer reduces the solvent’s thermodynamic quality towards the polymer that may cause the coil to shrink. The surface active monomer AAm adsorbs on the polymer coil and stabilizes the aggregates.[Citation15] In a such coil, the chain end will be found with high difficulty by the other chain end, and therefore the termination rate constant kt will be lower. Therefore, the reduction in solvent quality could result in an increased rate of polymerization in the presence of aggregated polyAAms. Indeed, the polyAAm chains are known to associate intermolecularly already at low conversion and build up a transitory network with a high viscosity.[Citation5] Under such conditions, the abrupt increase in the polymerization rate could appear.
Figure and Table show that the rate of AAm polymerization (Rp, max1) increases with increasing DAR concentration:(1)
Table 1. Variation of kinetic parameters with the DAR concentration in the photoinitiated micellar polymerization of AAm.
The reaction order x close to 0.5 indicates that the bimolecular termination governs the polymerization of AAm. This means that the compartmentalization of reaction loci (into particles) is not so effective.
The effect of AAm concentration on the photoinduced polymerization of AAm is summarized in the Figures and and Table . The final (limiting) conversion increases with increasing AAm concentration. The mixed hydrophobic Tw85/AAm micelles might contribute to the whole conversion and the limiting conversion as well.
Table 2. Variation of kinetic parameters with the DAR concentration in the photoinitiated micellar polymerization of AAm (SDS runs) with AAm concentration.
Figure 9. Variation of the rate of photoinduced polymerization of AAm with conversion and AAm concentrations. Recipe: 27 g H2O, 0.504 g Tw85, 0.103 g DAR (1) 0.5 g AAm, (2) 1.0 g AAm, (3) 1.5 g AAm, (4) 2.0 g AAm.
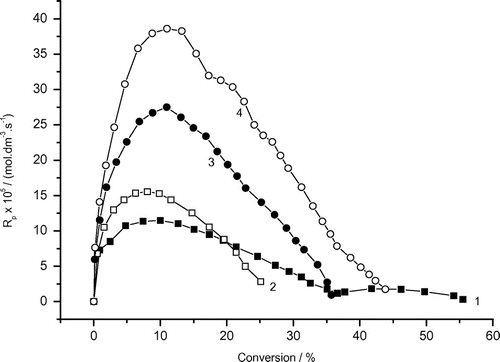
The dependence of the rate of polymerization vs. conversion listed in Figure is described by a curve with the rate maximum. This behavior deviates from the above McP of AAm in the presence of hydrophilic Tw60 emulsifier and the solution polymerization of acrylic ester monomers (Figure ) as well. The absence of the second rate maximum probably results from the glassy-state interval due to which the initiator DAR was trapped in the hydrophobic core of Tw85 micelles. Figure and Table show that the rate of AAm McP (Rp, max1) increases with increasing AAm concentration as follows:(2)
The apparent reaction order x = 1.4 might result from association/binding between AAm and polyAAm.
The McP of AAm with hydrophobic emulsifier (Tw85) micelles is somewhat slower than that with hydrophilic emulsifier (Tw60) micelles and both are faster than that without emulsifier (blank):(3)
This can be accompanied by the compartmentalization of reaction loci due to which the rate of polymerization increases. The largest synergistic effect of surface active polar AAm (which can act as a co-emulsifier [Citation15]) appears in the presence of emulsifier and the contribution of particular polymerization is larger with hydrophilic emulsifier. The increased location of hydrophobic DAR in the hydrophobic Tw85 micelles might increase the self-quenching and quenching of DAR* by AAm.
The initiating radicals are formed by decomposition of excited states of photoinitiator DAR*. The single radicals derived from DAR* are supposed to be in both micelles and aqueous phase. Similar polymerization behavior of both solution and McPs indicates that the intra- and intermolecular associates of polyacrylamide chains take place and they play the role of the compartmentalized reaction loci which are responsible for the complex variation of Rp with conversion. The quenching of DAR* by monomer and nonradiative deactivation of DAR* disfavor the formation of radicals and slow down the polymerization.[Citation20]
The mechanism of photophysical and phochemical processes proceeding in the present system can be summarize as follows (Figure ):(4)
(5)
(6)
(7)
Figure 10. Photochemical mechanism of primary radicals formation.
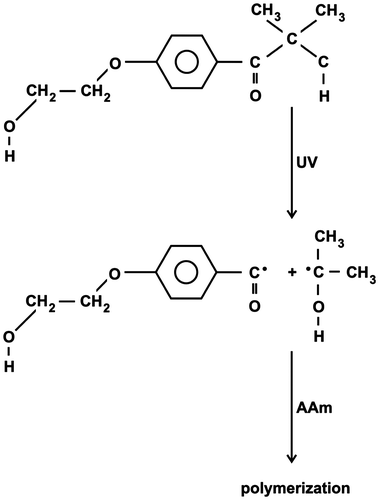
The decomposition of excited DAR* promotes the formation of initiating RAD•fr radicals resposible for the start of polymerization (Figure ) and the appearance of first rate maximum. The reaction of initiating radicals derived from DAR* with oxygen is expected to decrease the radical concentration and leads to the formation of the inactive (peroxide) products. The appearance of the second rate maximum probably indicate that there is a second source of radicals. These radicals can be formed by the decomposition of the excited {DAR…oxygen or peroxide product}*. Furthernore, the formation of insoluble polyAAm polymers indicates the formation of polymer gels or gelation. Thus, the gel effect could also contribute to the increase in the rate of polymerization at high conversions. The biradicals formed from the excited carbonyl groups in both DAR and polyAAm might form the cross-linked polymer gel.
There are several approaches concerning of the aqueous polymerization of AAm. Under the ideal solution polymerization, the long stationary-state rate interval is supposed to appear. This is not the case when the polymerization is accompanied by the self-association of polar monomers (e.g. acrylamide, acrylic acid,…) as well as their association with their polymer molecules (a template polymerization). Under such conditions, the dependence of rate of polymerization on conversion is much more complex. The polyAAm chains are known to associates themselves and with its monomer as well.[Citation10] The intra- and intermolecular association of polyAAm chains probably form nanospheres able to absorb AAm which then act as active nanoparticles. The formation of cross-linked polyAAm gels might result from the intermolecular association which induces the intra- and intermolecular cross-linking. The reaction order x (=1.4) might result from above-mentioned associations and cross-linking reactions.
The kinetic data of AAm polymerization with and without nonionic emulsifier might be also discussed in terms of hydrophobic and electrostatic interactions. Monomer (or oligomer) free radical (), which is polar (or has both positive and negative charges), associates/binds with polymer to give an inactive species:
(8)
where DARfr is the fraction of photoinitiator built into the monomer or oligomer species. This will result in the decrease of both monomer and initiating radical which leads to the decrease of the rate of polymerization. The hydrophobic interactions are responsible for association/binding between emulsifiers, and DAR species. A slower polymerization of AAm with hydrophobic emulsifier Tw85 is in agreement with step (8) due to which decreases the concentration of radicals derived from DAR* and AAm concentration.
Conclusion
The solution and McPs of AAm initiated by UV-light in the presence of the DAR photoinitiator were studied. Variations of kinetic parameters with DAR and AAm concentrations and with and without Tween emulsifiers were studied. Furthermore, the kinetics of AAm polymerization under argon and air atmospheres were investigated. The polymerization rate vs. conversion curve of the photoinduced polymerization of AAm was described by two and four nonstationary rate intervals. The rate of polymerization (Rp, max1, the first maximum rate) was observed to increase with both monomer and phoinitiator concentrations. The second rate maximum seems to appear as a result of accumulation of second source of radicals (peroxides) during the polymerization and gel formation. The intra- and intermolecular association and cross-linking of polyAAms and saturation of polyAAm associate with AAm transfer of the homogeneous polymerization system into the particular one.
Funding
This research is supported by the VEGA [project number 2/0037/10], [project number 2/0152/13]; and the APVV [project 0125-11].
References
- Segurola J, Allen N, Edge M, Roberts I. Photochemistry and photoinduced chemical crosslinking activity of acrylated prepolymers by several commercial type I far UV photoinitiators. Polym. Degrad. Stab. 1999;65:153–160.
- Capek I. Photopolymerization of butyl acrylate microemulsions 1. Post-polymerization. Polym. Int. 1996;40:41–49.
- Capek I, Potisk P. Microemulsion and emulsion polymerization of butyl acrylate. I. Effect of the initiator type and temperature. Eur. Polym. J. 1995;31:1269–1277.
- Capek I, Funke W. Kinetic investigation of the dispersion polymerization of N,N′-methylenebis(acrylamide) in the presence of various emulsifiers. Angew. Makromol. Chem. 1990;191:2549–2565.
- Rosen MJ. Surfactants and interfacial phenomena. 3rd ed. Hoboken (NJ): Wiley-Interscience, Wiley; 2004.
- Shukla JS, Mishra DC. Aqueous polymerization of acrylamide initiated by acidic potassium permanganate–ascorbic acid redox system. J. Polym. Sci. Polym. Chem. 1973;11:751–762.
- Kim OK. The catalytic role of micelle-bisulfite complexation in vinyl polymerization. In: Mittal KL, editor. Micellization, solubilization and microemulsions. Vol. 2. New York: Plenum Press; 1967. p. 627–644.
- Bartoň J, Juraničová V, Vašková V. Aqueous polymerization of vinyl monomers in the presence of surfactants. Angew. Makromol. Chem. 1985;186:1935–1941.
- Vašková V, Juraničová V, Bartoň J. Aqueous polymerization of vinyl monomers in the presence of surfactants IV. Effect of the chain length of the surfactant on the acrylamide polymerization. Chem. Pap. – Chem. Zvesti. 1986;40:435–441.
- Carver MT, Dreyer U, Knoesel R, Candau F. Kinetics of photopolymerization of acrylamide in AOT reverse micelles. J. Polym. Sci., Part A: Polym. Chem. 1989;27:2161–2177.
- Capek I. Photopolymerization of alkyl(meth)acrylates and polyoxyethylene macromonomers in fine emulsions. Eur. Polym. J. 1999;36:255–263.
- Capek I. On the free-radical microemulsion polymerization of butyl acrylate in the presence of poly(oxyethylene) macromonomer. Chem. Pap. – Chem. Zvesti. 1999;53:332–339.
- Tonnar J, Pouget E, Lacroix-Desmazes P, Boutevin B. Synthesis of poly(vinyl acetate)-block-poly(dimethylsiloxane)-block-poly(vinyl acetate) copolymers by iodine transfer photopolymerization in miniemulsion. Macromol. Symp. 2009;281:20–30.
- Chemtob A, Kunstler B, Croutxe-Barghorn C, Fouchard S. Photoinduced miniemulsion polymerization. Colloid. Polym. Sci. 2010;288:579–587.
- Candau F, Selb J. Hydrophobically modified polyacrylamides prepared by micellar polymerization. Adv. Colloid Interface Sci. 1999;79:149–172.
- Capek I, Janíčková S, Donescu D, Sarov Y, Rangelow IW. Microemulsion polymerization of butyl acrylate under ultrasound irradiation. Polym. J. 2006;38:1–13.
- Capek I. On photoinduced miniemulsion polymerization of butyl acrylate with clay. Des. Monomers Polym. 2012;15:345–355.
- Cardenas JN, O’Driscoll KF. High-conversion polymerization. I. Theory and application to methyl methacrylate. J. Polym. Sci. 1976;14:883–891.
- O’Driscoll KF. An analysis of the factors affecting the pressure dependence of the termination rate constant kt. Angew. Makromol. Chem. 1979;180:2053–2056.
- Timpe HJ. Photoinduced electron transfer I (Springer-Verlag, Berlin). Top. Curr. Chem. 1990;156:167–197.