
Abstract
The present study deals with the development and characterization of sorbitan monopalmitate (SM) organogels using mustard oil (MO) and groundnut oil (GO) as the apolar phase. In both the cases, the organogels containing ≥ 20% (w/w) SM were found to be stable for prolonged period. The microscopic studies showed the presence of aggregates of tubular shaped gelator fibers in the gel architecture. Fourier transform infrared spectroscopy indicated formation of intermolecular hydrogen bonding amongst the oil and gelator molecules. Differential scanning colorimetry studies indicated that the thermal stability of GO organogels was found to be more than the MO organogels. Gels showed pseudoplastic flow, with shear thinning and thixotropic behavior. The drug release from the organogels followed Higuchian kinetics with non-Fickian diffusion. The pH values of organogels were found to be near the human skin pH and highly hemocompatible. Metronidazole loaded organogels have shown good antimicrobial activity against Bacillus subtilis. Based on the preliminary studies, the developed organogels may be tried as drug carriers.
1. Introduction
Organogels may be defined as bi-continuous semi-solid formulations containing an apolar phase immobilized within the spaces available of a 3D networked structure of gelator molecules.[Citation1] Apolar phase may be of an organic solvent, mineral oil, and vegetable oil. Vegetable oils can be modulated to form organogels using nonionic surfactants (span 40, span 60) as the organogelators.[Citation2,Citation3]Sorbitan monopalmitate (span 40, SM) is a solid gelator and is an ester of sorbitol and palmitic acid.[Citation2] SM is solid at room-temperature but forms a clear solution at temperature >50 °C.[Citation2]
Mustard oil (MO) was being extracted from the seeds of mustard (Brassica juncea L.).[Citation4] MO primarily contains 1% of allyl isothiocyanate, 60% of monounsaturated fatty acids (MUFA), 21% of polyunsaturates, and 12% saturated fats. MUFA is generally composed of ~42% of euric acid and ~12% of oleic acid. The polyunsaturates are composed of 6% of ω-3 alpha-linolenic acid and 15% of ω-6 linoleic acid.[Citation5,Citation6] MO has low saturated fat content as compared to the other oils and has very good antioxidant and cholesterol reducing properties. In addition to the above, various essential vitamins (e.g. B vitamins and vitamin E) are also present in MO.[Citation6] It helps to avoid coronary heart diseases, detoxifies human body and stimulates digestion, circulation, and excretory system.[Citation7,Citation8]
Groundnut oil (GO) is the oil extracted from the seeds of groundnut (Arachis hypogaea L.).[Citation9] The major components of GO are fatty acids such as oleic acid, linoleic acid, and palmitic acid.[Citation10] It also contains antioxidants such as vitamin E.[Citation10,Citation11] Unsaturated fatty acids (e.g. oleic and linoleic acids) have permeation enhancing activity.[Citation12]
Keeping the facts in mind about the vegetable derived oils, it seems that the development of organogels using vegetable derived oils might add extra benefits during transdermal drug delivery. In addition to the pharmaceutical applications, the vegetable oil-based organogels may also find applications in food industry and cosmetic industries.[Citation13,Citation14] The gelation of the oil helps preventing migration of oils in composite pharmaceutical, food, and cosmetics products, thereby enhancing the shelf-life of the products.[Citation14,Citation15] The purpose of the current study is to develop vegetable oil (MO and GO) based organogels, which may be used as a matrix for topical/transdermal drug delivery.
2. Materials and methods
2.1. Materials
SM was purchased from Loba chemie, Mumbai, India. Edible MO and GO were purchased from the local market. Dialysis tubing (MW cutoff: 14 KDa) was purchased from Himedia, Mumbai, India. Metronidazole (MZ) was a kind gift from the Aarthi drugs Ltd Mumbai, India. All experimental studies were carried out using double distilled water (DDW).
2.2. Methods
2.2.1. Preparation of organogels
Accurately weighed SM was dissolved in specified amount of either MO or GO whose temperature was maintained at 60 °C and kept on stirring at 500 rpm. The solution was subsequently cooled to room temperature (RT) to achieve gelation. Gelation was considered success if the solution failed to flow under gravity upon inversion. The critical gelator concentration (CGC) was figured out by varying SM concentration. CGC may be defined as the minimum concentration of gelator required for immobilizing the apolar liquid.[Citation16]
Drug (MZ) loaded gels were prepared in a similar manner. Accurately weighed MZ was dissolved in the oil phase (either MO or GO) at 50 °C for 20 min. The drug solution in oil was used for the preparation of organogels. Apart from this, rest of the gel preparation was same as described above. The concentration of the drug within the organogels was maintained at 1% (w/w). All the samples were kept at RT for further analysis.
2.2.2. Organoleptic evaluation
The freshly prepared organogels were subjected to organoleptic evaluation by observing for their color, odour, taste, appearance, and texture.
2.2.3. Microscopic studies
A compound optical microscope (LEICA-DM 750 equipped with ICC 50-HD camera) was used for analyzing the microstructure of the samples. The mechanism of formation of organogels was studied by varying the proportions of SM in the oil.
2.2.4. Fourier transform infrared spectroscopy studies
Infrared spectroscopy of the samples was carried out by using a Fourier transform infrared (FTIR) spectrophotometer in attenuated total reflectance ATR mode (Alpha-E, Bruker, Germany). The raw materials and samples were scanned in the range of 3500 cm−1 to 500 cm−1 to understand the interactions amongst the components of the organogel.
2.2.5. XRD analysis
The samples were subjected to XRD analysis using Philips, XRD-PW 1700, Rockville, USA. Samples were scanned in the range of 10°–50° 2θ at a rate of 2° per min.
2.2.6. Accelerated thermal stability studies
The accelerated thermal stability test was carried out by thermo-cycling method.[Citation3] In short, freshly prepared organogel samples were subjected to repeated freezing-thawing cycles, by keeping them alternatively at –20 and 65 °C, respectively, for 15 min at each temperature. The study was continued for eight thermocycles. Samples were analyzed visually after every cycle for any destabilization. Organogels may be regarded as stable, if they can withstand at least five cycles of thermo-cycling.[Citation17,Citation18]
2.2.7. Thermal analysis
Falling ball method was used for predicting the gel to sol transition temperature (Tgs) of the organogels.[Citation19] A stainless steel (SS) ball of diameter 1/8th of an inch and weight 130 mg was used for the study. Accurately weighed 1 g of organogel was taken in the sample holder of melting point apparatus (EI-931, Mumbai, India). SS ball was placed on top of the sample and which was being maintained at 25 °C. The samples were then heated at 1 °C/min and the temperature at which the ball starts moving was noted as the Tgs of the organogel.[Citation19]
The organogels were subjected to differential scanning colorimetry (DSC) to analyze the thermal properties of the organogels using DSC-200 F3, MAIA, Netzsch, Germany over a temperature range of 25–90 °C at a heating rate of 2 °C/min. Accurately weighed sample (15–20 mg) was used for the analysis. Aluminum (Al) crucibles were used as the sample holders and were hermetically sealed with pierced Al lid.
2.2.8. Rheological studies
Controlled stress cone-plate viscometer (Bohlin visco 88, Malvern, UK) was used for the rheological properties of the organogels. All measurements were carried out using freshly prepared samples. A cone angle of 5.4° having a diameter of 30 mm was used. The gap between the cone and plate was maintained at 0.15 mm. The whole procedure was carried out at RT.
2.2.9. Spreadability studies
The spreadability of the organogels and marketed formulation (MZ1) was performed as per the reported method.[Citation20] In short, predetermined amount of samples (0.5 g) was placed in between two glass plates, of equal weight and area. The initial spreading diameter before placing the weight was noted. Thereafter, varying weights of 10, 20, 50, and 100 g were placed over the upper glass plate. The final diameter was noted over a period of 60 s. The spreadability of the formulations was reported as % spreadability, calculated as per the following equation.(1)
where D1, initial spreading diameter before loading; D2, final spreading diameter after loading.
2.2.10. Gel disintegration studies
Disintegration of organogels was carried out by using tablet disintegration apparatus (Electronics India, model: 901, India). For the study, 1 g of gel was placed in the disintegration test apparatus. DDW was used as the disintegrating media and maintained at 37 °C. The time taken for the complete disintegration of the gels, indicated by the absence of any component of the formulation in the disintegration basket was noted as the disintegration time.[Citation21]
2.2.11. pH measurement
The pH of organogels was measured by using digital ATC pH meter (EI instruments, model no: 132E, India). The pH of the organogels was measured by immersing the glass electrode of the pH meter in the samples.
2.2.12. Bulk resistance measurement
An in-house built DC impedance analyzer whose specification has been described in detail elsewhere was used to measure the bulk resistance of the organogels.[Citation3]
2.2.13. In vitro drug release studies
In vitro drug release studies were carried out using a modified Franz diffusion cell as described elsewhere.[Citation22–Citation24] Briefly, the donor and receptor of the diffusion cell was separated by a dialysis membrane. Accurately weighed 1 g of the organogel was taken in the donor chamber. DDW was used as the receptor medium. The receptor fluid was maintained at 37 ± 0.5 °C under constant stirring at 100 rpm. The whole receptor volume was changed periodically for every 15 min during first 1 h and then for every 30 min for the next 7 h. Aliquot of 5 ml was used for the spectrophotometric analysis at λmax 321 nm using UV–visible double-beam spectrophotometer (Labindia-3200, India).
2.2.14. Hemocompatibility studies
The hemocompatibility test was carried out to find out the extent of hemolysis in the presence of organogels. Goat blood was used for the study and the experiment was carried out as described elsewhere.[Citation3,Citation25] The % hemolysis was calculated as per the following formula:(2)
2.2.15. Antimicrobial studies
Antimicrobial assay was performed using bore well method.[Citation26] The antimicrobial efficiency of MZ incorporated organogels was studied against gram-positive bacteria B. subtilis. One ml of nutrient broth cell suspension containing 106–107 cfu/ml of bacteria was spread on solid nutrient agar plates using a sterilized spreader. Wells of diameter 5 mm were drilled in the plates using a sterilized borer so as to accommodate 0.2 g of sample. The plates were then incubated at 37 °C for 24 h and zone of inhibition was measured.
3. Results and discussion
3.1. Preparation of organogels
SM was dissolved in MO and GO at 60 °C, so as to obtain a clear homogenous solution. As the temperature of solution was subsequently lowered to RT, SM molecules started precipitating out of the oil phase due to the change in its solubility parameter.[Citation23,Citation27] The precipitated out molecules of SM self-assembled to form gelator fibers. The fibers, so formed, physical interacted amongst each other to form a 3D networked structure. During the cooling process, the hot clear solution of the gelator in oil phase becomes cloudy suspension. The cloudy suspension either remains cloudy if the concentration of the gelator molecules was lower than CGC or forms opaque gels if the concentration of the gelator molecules was higher than CGC (Figures and ). The gelation was considered successful, if the samples did not flow under gravity when the vials were inverted.[Citation28,Citation29] The CGC was found to be 18% (w/w) and 20% (w/w) for MO and GO, respectively. The MO gels containing 18% (w/w) SM were not stable after one month of storage at RT. GO gels obtained at CGC were found to be stable for more than one year. Hence to study the effect of gelator concentration and to compare the properties of the MO and GO organogels, organogels of different compositions were prepared as per Tables and . Equal gelator concentration was maintained for better comparison of both the systems.
Figure 1. SM/MO mixture (a) after melting, (b) below CGC and (c) above CGC.

Figure 2. SM/GO mixture (a) after melting (b) below CGC and (c) above CGC.

Table 1. Composition of the organogels used for further analysis.
Table 2. Composition of the drug containing organogels.
3.2. Organoleptic evaluation
The MG and GG organogels were found to be yellow and cream-white in color, respectively. As the concentration of the organogelator was increased, the consistency of the organogels improved. All the samples were found to be oily to touch and were having gritty nature. The organogels were having a bland taste.
3.3. Microscopic studies
The microstructures of the organogels were studied under light microscope (Figures and ). The micrographs suggested that the SM molecules precipitated out as tubular structures. As the concentration of the gelator was increased, there was a subsequently increase in the length of the tubules to form gelator fibers. With the subsequent increase in the gelator concentration, there was an increase in the density of the gelator fibers. At CGC, these fibers formed a 3D networked skeleton which helped in the immobilization of the MO and GO.[Citation30,Citation31] The formation of the networked structure may be attributed to the apolar–apolar interactions amongst the fatty acyl chains of gelator and oil molecules. Higher hydrophobic interactions amongst the gel components improved the stability of the organogels.[Citation1] At CGC of the MO organogel, the hydrophobic interactions might not be sufficient to maintain the structural integrity of organogels for longer duration of time which lead to syneresis of the gels within a short span of time. At higher concentrations of gelator (≥20%), the syneresis of the oil phase was not observed indicating an improved stability of the organogels due to the increase in the hydrophobic interactions.[Citation26]
Figure 3. Micrographs of SM/MO mixture at the SM concentration of: (a) 5% (w/w), (b) 10% (w/w), (c) 15% (w/w), (d) 20% (w/w), (e) 23% (w/w) and (f) 25% (w/w).

Figure 4. Micrographs of SM/GO mixture at the SM concentration of: (a) 5 % (w/w), (b) 10% (w/w), (c) 15% (w/w), (d) 20% (w/w), (e) 23% (w/w) and (f) 25% (w/w).

3.4. FTIR analysis
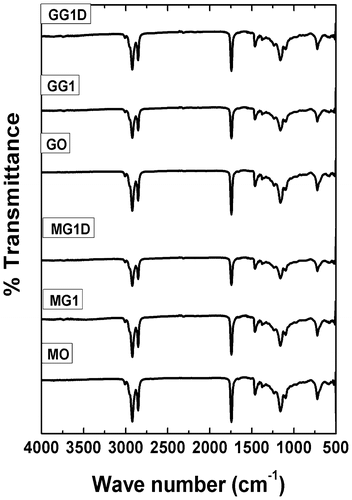
Figure shows the FTIR spectrograms of MO, GO, and organogels. FTIR spectral analysis gives information regarding interactions amongst the gel components. The spectral data of organogels was compared against the FTIR spectra of SM and MZ which were described elsewhere.[Citation3] FTIR spectrum of the SM showed a broad peak at 3300 cm−1 which denote the presence of O–H stretching vibrations.[Citation1,Citation32] The absence of the peak at 3300 cm−1 in the organogels suggested intermolecular hydrogen bonding amongst the fatty acyl groups of gelator and oil molecules.[Citation33] Formation of intermolecular hydrogen bonds might be responsible for the strength of the gels.[Citation34] Apart from this, there were no significant differences in the spectral pattern of the MO, MG, MGD, GO, GG, and GGD indicating that the chemical functionality of the gel components was conserved even after the addition of the drug. These results indicated that there is no interaction of the drug molecules with that of the gel components.
Figure 5. FTIR spectrograms of the samples.
3.5. XRD analysis
The XRD diffractograms of the organogels (MG1, MG1D, GG1, and GG1D) have been shown in Figure and their parameters have been given in Table . The area under the curve (AUC) and full width half maximum (FWHM) of the XRD profiles of the organogels suggested that the GO organogels were more amorphous than the MO organogels. It was observed that the drug containing organogels (MG1D, GG1D) showed lower AUC and FWHM values compared to their corresponding blank organogels (MG1 and GG1). The decrease in AUC and FWHM values suggested that MG1D and GG1D have higher crystallinity than MG1 and GG1, respectively. This may be attributed to the increased intermolecular hydrogen bonding.[Citation3,Citation18,Citation35]
Figure 6. X-ray diffractograms of the organogels.
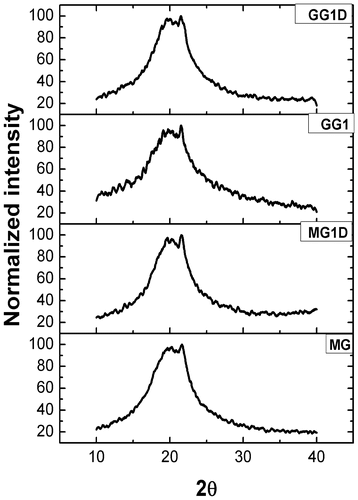
Table 3. XRD analysis of organogels.
3.6. Accelerated thermal stability studies
Accelerated thermal stability test was carried out by thermo-cycling method. The method involved incubation of the samples alternatively at higher (65 °C) and lower (–20 °C) temperatures. The study helps in predicting the mechanism of destabilization of the samples either due to the change in the physicochemical properties at the extreme conditions. At higher temperatures, the chances of oxidative changes are predominant which may alter the stability of the samples whereas at lower temperatures, the formation of solidified structures may alter the physical interactions amongst the sample components responsible for the formation of the networked structures. This method only gives a prediction about the stability. In general, it is expected that the samples should withstand at least five cycles of freeze-thaw process to be regarded as stable samples.[Citation17,Citation18,Citation36] All the samples (mentioned in Table ) were found to be stable for more than eight cycles.
3.7. Thermal analysis
The Tm (melting point) of the organogels was found by using falling ball method and have been tabulated in Table . The results indicated that with increase in the concentration of the gelator there was an increase in the Tm of both types of organogels.
Table 4. Tm values of the organogels by falling ball method.
Thermal profiles of the organogels were monitored using a differential scanning calorimeter. The results have been shown in Figure and tabulated in Table . The DSC studies indicated that the Tm value of GG organogels was higher than the MG organogels. Apart from Tm value, area under the melting endotherm (change in enthalpy, ΔH) was higher for the GG organogels when compared to the MG organogels. In addition to this, lower change in entropy (ΔS) values of GG organogels during melting also indicated that GG organogels were thermally more stable than the MG organogels (Table ). Higher Tm, ΔH, and lower ΔS values suggested that the GG organogels were more stable than the MG organogels.[Citation37,Citation38] Addition of drug did not cause any significant variation in the thermal properties of both the types of organogels.
Figure 7. DSC thermograms of the organogels.
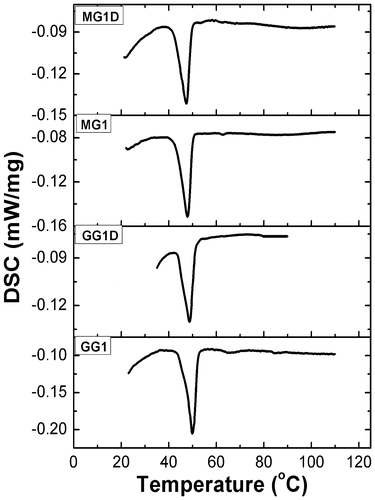
Table 5. DSC data of the organogels.
3.8. Rheological studies
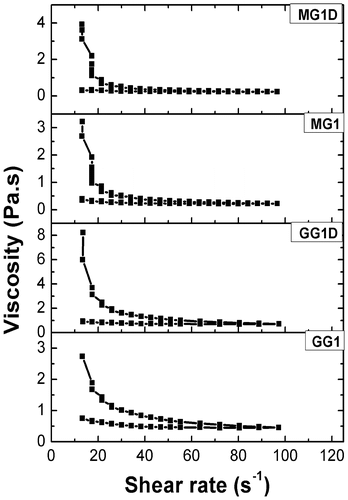
The viscosity profiles of the organogels have been shown in Figure . Viscosity profiles indicated that the apparent viscosity of the GG organogels were higher as compared to the MG organogels. Incorporation of the drug (MZ) into the organogels resulted in the higher apparent viscosity as compared to the blank gels. In general, the viscosity of organogels was lowered with increase in the shear rate. The organogels have shown higher apparent viscosities when the shear rate was increased from 12 to 95 s−1 and lower apparent viscosities when shear rate was decreased from the 95 to 12 s−1. Variation in the apparent viscosity at a particular shear rate leads to the formation of hysteresis loop.[Citation39] Decrease in viscosity with increase in shear rate and formation of hysteresis loop during cycle of shears indicated the shear thinning and thixotropic behavior of the organogels.[Citation40] During thixotropic behavior, both the organogels did not show 100% recovery of apparent viscosities but many junction points were recovered. This suggested that the total recovery of structural loss was not possible in these organogels. This kind of behavior is common in gels, ointments, and creams.[Citation41] The fluid flow behavior of the organogels was determined by fitting the data in modified power law (equation Equation3(3) ).[Citation42]
(3)
Figure 8. Viscosity profiles of the organogels.
where η is the viscosity (Pa.s) at shear rate, γ (s−1); K is the flow consistency index and n is the flow behavior index.
The values of n determine the type of fluid behavior. The n values of organogels were found to be less than 1 which indicated that the organogels were non-Newtonian fluids with pseudoplastic flow behavior (Figure and Table ). Both MG and GG gels did not lose their structural integrity even at high shear rates. Presence of shear thinning behavior with thixotropic properties is considered to be essential property for topical formulations.[Citation43] In addition to this, the semi-solid nature of MG and GG gels at low shear rates allow their long-term stability during storage.[Citation43,Citation44]
Figure 9. Graph between viscosity and shear rate (log–log scale) of the organogels.
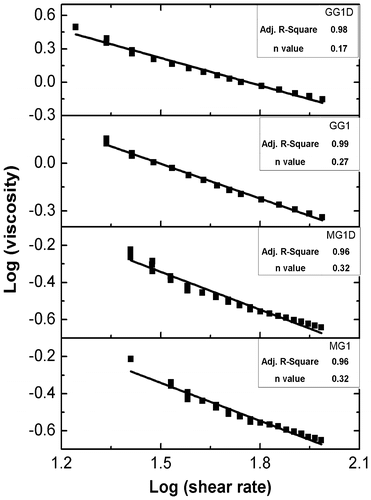
Table 6. Nature of the organogels based on the n values.
3.9. Spreadability studies
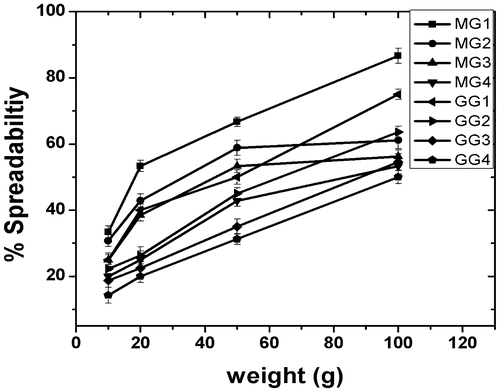
The % spreadability with respect to varying weights has been shown in Figure . There was an increase in the % spredability as the applied load was increased. As the gelator concentration was increased, the % spreadability decreased significantly, which may be attributed to the increase in physical strength of the organogels. Presence of dense 3D microstructure of organogelator at higher concentration resulted in the formation of stronger gels. The gels were found to be evenly spread without any phase separation. This indicated that the gels were having good structural integrity and may be used as a vehicle for topical and transdermal drug delivery.[Citation18,Citation45,Citation46] It was found that at equal gelator concentration, MG organogels were having higher % spreadability than the GG organogels. This may be attributed to the higher structural integrity as was evident from the thermal and viscosity results. Thermal studies indicated that the Tm of GG gels were higher as compared to the MG gels. Also, the apparent viscosity of the GG gels was higher than the MG gels. The results suggested that the GG gels had stronger interactions amongst the gel components which, in turn, resulted in their higher mechanical stability.[Citation20,Citation39]
Figure 10. Plot showing the % spreadability of organogels.
3.10. Disintegration studies
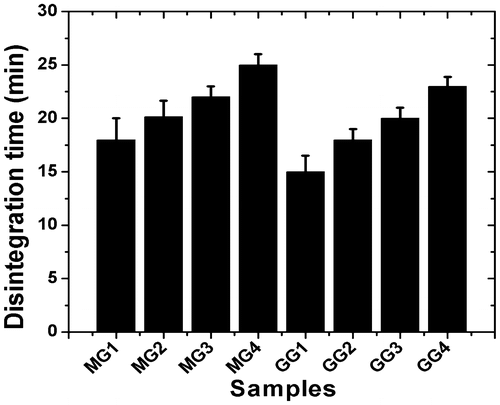
The disintegration of MG and GG gels was completed in 15–25 min (Table ). As the gelator concentration was increased, the disintegration time also increased in both the types of organogels. The increase in disintegration with increase in gelator concentration might be due to the increase in density of 3D gelator network within the organogels. The increase in gelator network density was evident from the microscopic studies. At equal gelator concentration, MG organogels have shown higher disintegration compared to GG organogels (Figure ). This may also be associated with the higher mechanical stability of the GG organogels.[Citation21]
Table 7. Disintegration times of the organogels.
Figure 11. Plot showing disintegration time of organogels.
3.11. Bulk resistance measurement of organogels
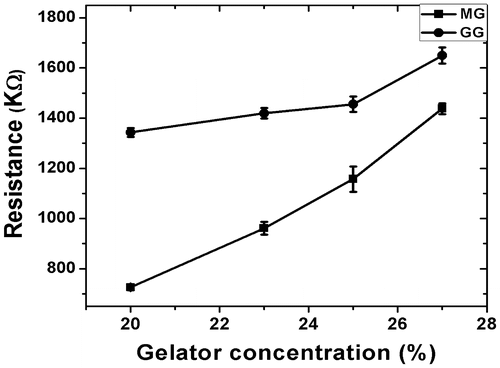
The measurement of the bulk resistance suggested that the impedance of the gels increased with the increase in the gelator concentration (Figure ). The bulk resistance of the MG organogels was lower as compared to the GG organogels. This may be associated to the higher hydrophobic interactions amongst the GG organogel components.
Figure 12. Plot of gelator concentration vs resistance of organogels.
3.12. In vitro drug release studies
Release of drug molecules from formulations depend upon the solubility and partition coefficient of the drug in oil. The in vitro drug delivery studies were carried out using 1% (w/w) MZ loaded organogels. Depending on the composition of the organogels, the cumulative percentage drug release (CPDR) of the MZ was found to be in between 60 and 70% at the end of 12 h (Figure ). The increase in gelator concentration in the organogels decreased the release of the drug molecules of both the types (MG and GG) of organogels. This may be attributed to the formation of dense networked structures as the gelator concentration was increased. The increase in the density of the gelator network hinders the diffusion of the drug molecules.[Citation47,Citation48] The drug release was higher in the MG gels as compared to the GG gels, when the composition of both the gels was kept constant. This may be associated with the formation of more compact structure in GG gels. This resulted in the greater hindrance of the diffusion of the drug molecules (Table ).[Citation49]
Figure 13. CPDR profile for different compositions of the (a) MG and (b) GG organogels as a function of time.
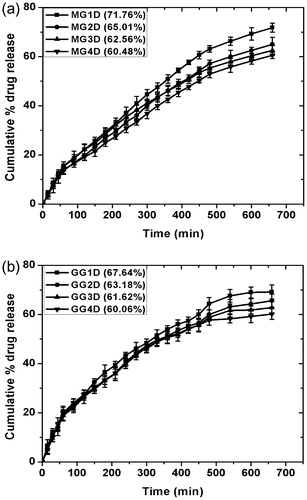
Table 8. Kinetics of in vitro drug release.
3.13. pH measurement
The pH values of the organogels were found to be in the range of 5.3 and 6.3 (Table ). This suggested that the developed organogels may not cause skin irritation. Hence, they may be applied over skin without any side effects.[Citation3,Citation39,Citation50]
Table 9. pH values of organogels.
3.14. Hemocompatibility test
The % hemolysis of goat RBCs was measured and has been tabulated in Table . The results suggested that all the organogel samples were highly hemocompatible (% hemolysis <5%) and may be regarded as biocompatible in nature.[Citation25,Citation51,Citation52]
Table 10. % hemolysis of the organogels.
3.15. Antimicrobial studies
The results of antimicrobial efficiency of the developed and the marketed formulations have been tabulated in Table . MG1 and GG1 served as negative control while MZ was used as positive control. The antimicrobial activity of MG1D, GG1D against B. subtilis was found to be significant and the results were comparable to marketed formulation. MG1D organogels were having higher zone of inhibition than the GG1D organgels. The blank gel did not show any antimicrobial activity, suggesting that the zone of inhibition was due to the incorporated drug only.
Table 11. Zone of inhibition obtained against B. subtilis.
4. Conclusion
In the present study, MO- and GO-based organogels were successfully developed. SM was used as an organogelator. The organogels were found to be opaque, semi-solid, thermo-reversible pseudoplastic, shear thinning, and thixotropic in nature. The organogels were thermally stable over a wide range of temperature. DSC studies indicated that GG gels showed higher Tm as compared to MG gels indicating that the GG gels were thermally more stable. The gelator concentration showed a direct influence upon the gel disintegration. The drug release from the organogels was found to follow Higuchian kinetics with non-Fickian diffusion. The drug loaded gels showed good antimicrobial efficiency against B. subtilis. The gels were found to be highly hemocompatible. The preliminary studies suggested that the developed organogels have huge potential to be used as controlled delivery matrices.
Acknowledgement
The authors acknowledge the financial and instrumental facilities provided by the National Institute of Technology, Rourkela, India. The funds leveraged from the project (BT/PR14282/PID/06/598/2010) sanctioned by the Department of Biotechnology, Govt. of India is hereby acknowledged.
References
- Vintiloiu A, Leroux JC. Organogels and their use in drug delivery — A review. Journal of Controlled Release. 2008;125:179–192.
- Murdan S, Gregoriadis G, Florence AT. Novel sorbitan monostearate organogels. Journal of Pharmaceutical Sciences. 1999;88:608–614.
- Behera B, Patil V, Sagiri SS, Pal K, Ray SS. Span-60-based organogels as probable matrices for transdermal/topical delivery systems. J. Appl. Polym. Sci. 2011;125:852–863.
- Mustorp S, Engdahl-Axelsson C, Svensson U, Holck A. Detection of celery (Apium graveolens), mustard (Sinapis alba, Brassica juncea, Brassica nigra) and sesame (Sesamum indicum) in food by real-time PCR. European Food Research and Technology. 2008;226:771–778.
- Yu JC, Jiang Z, Li R, Chan SM. Chemical composition of the essential oils of Brassica juncea (L.) Coss. grown in different regions, Hebei, Shaanxi and Shandong, of China. J. Food Drug Anal. 2003;11:22–26.
- Kaushik N, Agnihotri A. GLC analysis of Indian rapeseed-mustard to study the variability of fatty acid composition. Biochemical Society Transactions. 2000;28:581–582.
- Kumar S, Pahujani S, Ola RP, Kanwar SS, Gupta R. Enhanced Thermostability of Silica-immobilized Lipase from <i>Bacillus coagulans</i> BTS-3 and Synthesis of Ethyl Propionate. Acta microbiologica et immunologica hungarica. 2006;53:219–231.
- Fox LT, Gerber M, Plessis JD, Hamman JH. Transdermal Drug Delivery Enhancement by Compounds of Natural Origin. Molecules. 2011;16:10507–10540.
- Anyasor G, Ogunwenmo KO, Oyelana OA, Ajayi D, Dangana J. Chemical analyses of Groundnut (Arachis hypogaea) oil. Pak. J. Nutr. 2009;8:269–272.
- Dandavate V, Jinjal J, Keharia H, Madamwar D. Production, partial purification and characterization of organic solvent tolerant lipase from Burkholderia multivorans V2 and its application for ester synthesis. Bioresource Technology. 2009;100:3374–3381.
- Wang K, Jia Q, Han F, Liu H, Li S. Self-assemble l-alanine derivative organogel as in situ drug delivery implant: characterization, biodegradability, and biocompatibility. Drug Delivery Ind. Pharm. 2010;36:1511–1521.
- Aungst BJ. Structure/effect studies of fatty acid isomers as skin penetration enhancers and skin irritants. Pharmaceutical Research. 1989;06:244–247.
- Bot A, den Adel R, Roijers EC. Fibrils of γ-Oryzanol + β-Sitosterol in Edible Oil Organogels. Journal of the American Oil Chemists' Society. 2008;85:1127–1134.
- Wright AJ, Marangoni AG. Formation, structure, and rheological properties of ricinelaidic acid-vegetable oil organogels. Journal of the American Oil Chemists' Society. 2006;83:497–503.
- Kodali DR. The utilization of rice bran wax to stabilize long chain ω-3 polyunsaturated fatty acid esters. Lipid Technology. 2009;21:254–256.
- Kumar R, Katare OP. Lecithin organogels as a potential phospholipid-structured system for topical drug delivery: a review. Aaps Pharmscitech. 2005;6:E298–E310.
- Takafuji M, Azuma N, Miyamoto K, Maeda S, Ihara H. Polycondensation and Stabilization of Chirally Ordered Molecular Organogels Derived from Alkoxysilyl Group- Containing l -Glutamide Lipid †. Langmuir. 2009;25:8428–8433.
- Sagiri SS, Behera B, Sudheep T, Pal K. Effect of Composition on the Properties of Tween-80 Span-80-Based Organogels. Designed Monomers & Polymers. 2012;15:253–273.
- Lin KY, Wang DM, Lai JY. Nonsolvent-induced gelation and its effect on membrane morphology. Macromolecules. 2002;35:6697–6706.
- Jadhav KR, Kadam VJ, Pisal SS. Formulation and evaluation of lecithin organogel for topical delivery of fluconazole. Curr. Drug Delivery. 2009;6:174–183.
- Mukhopadhyay P, Fujita N, Takada A, Kishida T, Shirakawa M, Shinkai S. Regulation of a Real-Time Self-Healing Process in Organogel Tissues by Molecular Adhesives. Angewandte Chemie International Edition. 2010;49:6338–6342.
- Samanta M, Dube R, Suresh B. Transdermal drug delivery system of haloperidol to overcome self-induced extrapyramidal syndrome. Drug Delivery Ind. Pharm. 2003;29:405–415.
- Upadhyay KK, Tiwari C, Khopade AJ, Bohidar HB, Jain SK. Sorbitan ester organogels for transdermal delivery of sumatriptan. Drug Delivery Ind. Pharm. 2007;33:617–625.
- Masotti A, Ortaggi G. Chitosan Micro- and Nanospheres: Fabrication and Applications for Drug and DNA Delivery. Mini-Reviews in Medicinal Chemistry. 2009;9:463–469.
- Dutta P, Dey J, Perumal V, Mandal M. Amino acid based amphiphilic copolymer micelles as carriers of non-steroidal anti-inflammatory drugs: solubilization, in vitro release and biological evaluation. Int. J. Pharm. 2011;407:207–216.
- Bhattacharya C, Kumar N, Sagiri SS, Pal K, Ray SS. Development of span 80–tween 80 based fluid-filled organogels as a matrix for drug delivery. J. Pharm. Bioallied Sci. 2012;4:155.
- Yu H, Shi K, Liu D, Huang Q. Development of a food-grade organogel with high bioaccessibility and loading of curcuminoids. Food Chem. 2011;131:48–54.
- Gupta M, Vaidya B, Mishra N, Vyas SP. Effect of surfactants on the characteristics of Fluconazole Niosomes for enhanced cutaneous delivery. Artif. Cells, Blood Substitutes, Biotechnol. 2011;39:376–384.
- Pardakhty A, Shakibaie M, Daneshvar H, Khamesipour A, Mohammadi-Khorsand T, Forootanfar H. Preparation and evaluation of niosomes containing autoclaved Leishmania major: a preliminary study. J. Microencapsulation. 2011;29219–224.
- Namdeo A, Jain N. Liquid crystalline pharmacogel based enhanced transdermal delivery of propranolol hydrochloride. Journal of Controlled Release. 2002;82:223–236.
- Murdan S., Organogels in drug delivery. Expert Opin. on Drug Delivery. 2005;2:489–505.
- Shirakawa M, Kawano S, Fujita N, Sada K, Shinkai S. Hydrogen-bond-assisted control of H versus J aggregation mode of porphyrins stacks in an organogel system. Journal of Organic Chemistry 2003;68:5037–5044.
- Dautel OJ, Robitzer M, Lère-Porte J-P, Serein-Spirau F, Moreau JJ. Self-organized ureido substituted diacetylenic organogel. Photopolymerization of one-dimensional supramolecular assemblies to give conjugated nanofibers. Journal of the American Chemical Society 2006;128:16213–16223.
- Li Y, Wang T, Liu M. Ultrasound induced formation of organogel from a glutamic dendron. Tetrahedron. 2007;63:7468–7473.
- Dassanayake LSK, Kodali DR, Ueno S. Formation of oleogels based on edible lipid materials. Curr. Opin. Colloid Interface Sci. 2011;;16:432–439.
- Hussein MA, Asiri AM. Brill online books and journals. Des. Monomers Polym. 2012;15
- George M, Weiss RG. Chemically Reversible Organogels via “Latent” Gelators. Aliphatic Amines with Carbon Dioxide and Their Ammonium Carbamates †. Langmuir. 2002;18:7124–7135.
- Bauer T, Thomann R, Mülhaupt R. Two-component gelators and nucleating agents for polypropylene based upon supramolecular assembly. Macromolecules. 1998;31:7651–7658.
- Pénzes T, Csóka I, Erős I. Rheological analysis of the structural properties effecting the percutaneous absorption and stability in pharmaceutical organogels. Rheologica Acta. 2004;43:457–463.
- Terech P, Pasquier D, Bordas V, Rossat C. Rheological properties and structural correlations in molecular organogels. Langmuir. 2000;16:4485–4494.
- Mercurio DJ, Spontak RJ. Morphological Characteristics of 1,3:2,4-Dibenzylidene Sorbitol/Poly(propylene glycol) Organogels. Journal of Physical Chemistry B. 2001;105:2091–2098.
- Le Le Pluart L, Duchet J, Sautereau H, Halley P, Gerard J-F. Rheological properties of organoclay suspensions in epoxy network precursors. Applied Clay Science 2004;25:207–219.
- Lim PF, Liu XY, Kang L, Ho PC, Chan SY. Physicochemical effects of terpenes on organogel for transdermal drug delivery. International Journal of Pharmaceutics. 2008;358:102–107.
- Andrews GP, Jones DS. Rheological characterization of bioadhesive binary polymeric systems designed as platforms for drug delivery implants. Biomacromolecules. 2006;7:899–906.
- Dassanayake LSK, Kodali DR, Ueno S, Sato K. Physical properties of rice bran wax in bulk and organogels. J. Am. Oil Chem. Soc. 2009;86:1163–1173.
- Rane I, Thorat SP, Smita M. Formulation and in vitro evaluation of lecithin (soya and egg) based aceclofenac organogels. J. Pharm. Res. 2010;3:1438–1441
- Gander B, Gurny R, Doelker E, Peppas NA. Effect of polymeric network structure on drug release from cross-linked poly (vinyl alcohol) micromatrices. Pharmaceutical Research. 1989;06:578–584.
- Gander B, Beltrami V, Gurny R, Doelker E. Effects of the method of drug incorporation and the size of the monolith on drug release from cross-linked polymers. Int. J. Pharm. 1990;58:63–71.
- Ruiz M, Clares B, Morales ME, Gallardo V. Preparation, rheological study, and characterization of an organogel as a system for transdermal release of active principles. Pharmaceutical Development and Technology. 2007;12:637–644.
- Fu X, Yang Y, Wang N, Wang N, Yang Y. A novel chiral separation material: polymerized organogel formed by chiral gelators for the separation ofD- andL-phenylalanine. J. Mol. Recognit. 2007;20:238–244.
- Azeem A, Anwer MK, Talegaonkar S. Niosomes in sustained and targeted drug delivery: some recent advances. J. Drug Target. 2009;17:671–689.
- Janes K, Calvo P, Alonso M. Polysaccharide colloidal particles as delivery systems for macromolecules. Advanced Drug Delivery Reviews. 2001;47:83–97.