
Abstract
In the field of functional polymers, new aromatic polyethers were obtained with good yields by interfacial polycondensation of (EBr2)n and bisphenol A (bis(4-hydroxyphenylether) or catechol (2-hydroxyphenol) with various catalysts (Aliquat, TBAB, and TDA1).The structure of all aromatic polyethers was determined by means of 1H and 13C NMR, and infrared spectra. All polyethers had good solubilities and could be soluble in usual solvents such as acetone, chloroform, and dichloromethane. Average molecular weight ranged from 1000 to 11000 g mol−1. Their thermal characteristics were also studied by differential scanning calorimetry. The hydrolysis of these aromatic polyethers esters gives aromatic polyethers acids.
1. Introduction
Synthesis of functional and reactive polymers has received much attention since they can be used in many applications as supports, catalysts, or reagents for various reactions.[Citation1,2] Two methods exist for the synthesis of these polymers. The first one is based on polymerization of monomers containing the desired functional group. The second one is based on chemical modification of performed polymers.[Citation3,4]
In this work, we propose the synthesis of new functional polyethers acids based on α,α′- dibrominated diesters (EBr2)n (n being the number of methylene units) by phase transfer catalysis followed by hydrolysis. These polymers can find many applications, for instance as tensioactives agents,[Citation5] ion exchangers [Citation6,7], and ion sensitive field effect transistors [Citation8,9] (ISFET). The synthesis and the physical properties are described in this paper.
2. Experimental section
2.1. Materials
Bisphenol A, BPA, (bis(4-hydroxyphenyl)ether), and catechol (2-hydroxyphenol) (Aldrich) were purified by recrystallization from toluene. Nitrobenzene, chlorobenzene, and toluene (fluka) were purified by distillation under reduced pressure. Phase transfer catalysts: Tricaprylmethylammonium chloride (Aliquat), Tris(dioxa-3,6-heptyl)amine (TDA1), and tetrabutylammonium bromide(TBAB) were used without further purification. Adipic, pimelic, suberic, azelaic, and sebacic acids were purified by recrystallization from toluene.
2.2. Synthesis of dibromodiesters (EBr2)n, (e.g. n = 6)
Dimethyl-α,α′-dibromosebaçate ester was obtained [Citation10,11] by the action of thionyl chloride (0.71 g, 6 mmol) on sebacic acid (0.56 g, 2.8 mmol) to give the corresponding diacid chloride (sebaçoyl dichloride). Then bromation in α position of the carbonyl groups is performed followed by esterification with methanol (Figure ).
Figure 1. Synthesis of dibromo diesters (EBr2)n.
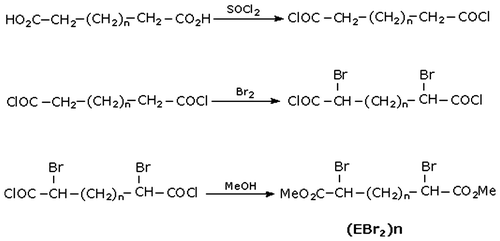
The yield of the product was 86 %. FTIR (CCl4): 2930, 2850, 1740, 1430, 1270, 1020, 785 cm−1. 1H NMR (δ, (ppm) from TMS in CDCl3), 1.3 and 2 (2m not separated, 12H, –CH2–), 3.7 (s, 6H, –COOCH3), 4.3 (t, 2H, –CHBr–). Analysis calculated for C12H20O4Br2 gave C 37.13, H 5.19; found C 37.80, H 5.19.
2.3. Phase transfer catalyzed two-phase polycondensation (e.g. polym P6b)
The polyethers were synthesized by polycondensation of (EBr2)n with BPA or catechol using phase transfer catalyst. In a typical polycondensation reaction, 5 mmol of bisphenol and 5 mmol of K2CO3were stirred with 1 mmol of aliquatas catalyst. To the stirred mixture was added a solution of dibromo derivative (EBr2)6 (1.94 g, 5 mmol) in 20 mL of nitrobenzene. (Figure ).
Figure 2. Synthesis of polyether.
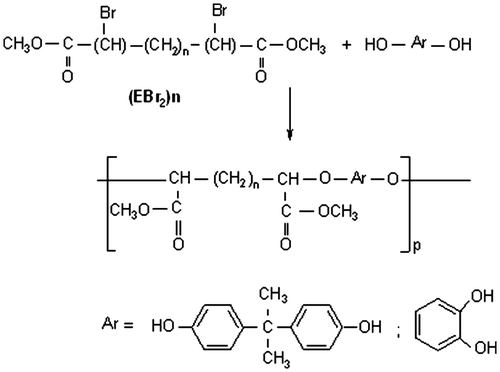
The two-phase mixture was vigorously stirred at the desired temperature for 4 h under nitrogen atmosphere. The reaction mixture was then cooled to room temperature and acidified with hydrochloric acid. The organic phase was poured into an excess of methanol and the polyethers, thus precipitated, were isolated, and dried under vacuum. The term ‘yield’ will be used in this work to express the amount of material obtained following these operations.
- Polyether from catechol and (EBr2)6
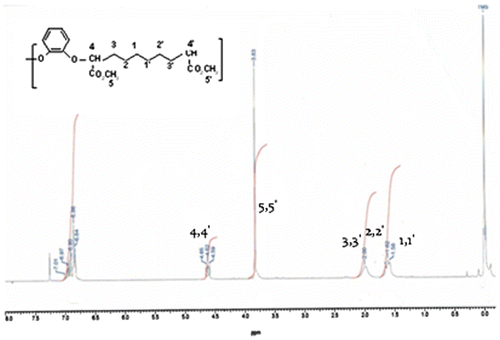
FTIR(film, cm−1): 2930(–CH2–), 1730(C=O, ester), 1595(CAr = CAr), 1263(-O-Ar). 1H NMR (δ, (ppm) from TMS in CDCl3) (Figure ) = 1,58[m, 4H, –CHO–CH2–CH2–CH2–CH2–CH2–CH2–CH–O–]; 1,62[m, 4H, –CHO–CH2–CH2–CH2–CH2–CH2–CH2–CH–O–];2,00[m, 4H, –CHO–CH2–CH2–H2–CH2–CH2–CH2–CH–O–]; 3,83 [s, 6H,COOCH3]; 4,62 [t, 2H, –CHO–]; 6,7–6,9[ m, 8H, –O–C6H4O–].
Figure 3. 1H NMR spectrum of polymer 6 g.
-Polyetherfrom BPA and (EBr2)6
FTIR(film, cm−1): 2933 (–CH2–, –CH3), 1731 (C=O, ester), 1233 (–O–Ar), 1600 (CAr = CAr).
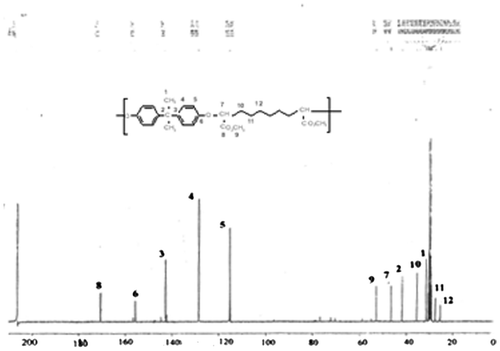
1HNMR(δ, (ppm) from TMS in CDCl3) = 1,3 and 2 [2 m, 12H, –CH2–]; 3,8 [s, 6H,-COOCH3]; 4,7 [t, 2H, –CH–O–Ar–]; 1,6 [s, 6H, –Ar–C(CH3)2–Ar–]; 6,8 and 7,2 [2d, 6H, –O–Ar–C(CH3)2–Ar–O–]. 13C NMR(δ, (ppm) from TMS in CDCl3) (Figure ) = 25; 27; 30; 31; 41,8; 46; 52; 115; 128; 142; 155; 172.
Figure 4. 13C NMR spectrum of polymer 6c.
2.4. Hydrolysis of polyethers with esters groups
In a 50 mL flask were placed 1 g of polyether ester and 0.04 g of sodium hydroxide dissolved in 10 ml of water and the mixture was refluxed for 2 h until total dissolution of polymer. The solution was cooled then acidified with hydrochloric acid and the polyether acid precipate. The product was filtered and dried under reduced pressure at 60 °C.
-Hydrolysis of polyethers obtained from catechol
1HNMR(δ, (ppm) from TMS in CDCl3) = 1.3 and 1.7 [2 m not separated, 8H, –CHO–CH2–CH2–CH2–CH2–CH2–CH2–CH–O–]; 1.9 [m, 4H, –CHO–CH2– (CH2)4–CH2–CH–O–]; 4.5 [t, 2H, –O–CH–(CH2)6–CH–O–]; 6.9 [m, 8H, –O–Ar–O]; 10.2[m,2H, –OCOH]
-Hydrolysis of polyethers obtained from BPA
1HNMR(δ, (ppm) from TMS in CDCl3) = 1.4 and 1.9 [2 m not separated, 12H, -CH2-]; 1.6 [s, 6H, –Ar–C(CH3)2–Ar];4.6 [t, 2H, –Ar–O–CH–(CH2)6–CH–O–Ar]; 7 [m, 8H, –Ar–(CH3)2–Ar];10.2 [m, 2H, –OCOH]
2.5. Characterization methods
1H NMR spectra were recorded on a Bruker AM-200 spectrometer (France). Infrared spectra were recorded with a BOMEM spectrometer (France). Size exclusion chromatography (SEC) was performed on an automated Waters HPLC/SEC system (France) with refractive index detector, operated at 30 °C using tetrahydrofuran as the eluting solvent at a flow rate of 0.8 mL.mn−1 and standard polystyrene as a reference. Transition temperatures were recorded with a Setaram 92 (France) differential scanning calorimeter at a heating rate of 10 °C min−1. The Tg was taken as the onset temperature of shift from the baseline.
3. Result and discussion
3.1. Synthesis of polyether
The general polycondensation reaction scheme related to this investigation is:
Phase transfer catalysis is a very convenient method for the synthesis [Citation12–14] or the chemical modification of polymers [Citation15,16] and phenols are particularly well suited for interfacial polycondensation [Citation17] because of their acidic character. All polyethers were synthesized by interfacial polycondensation of BPA or catechol with different (EBr2)n.
The reactions were carried out under solid–liquid phase transfer catalysis because in the presence of water the ester (EBr2)n can be hydrolyzed, and give the corresponding salt, which is low soluble and has a low reactivity in the organic phase. For the same reason, K2CO3 was used instead of KOH or NaOH to avoid the hydrolysis of ester.
The polyethers were purified by precipitation and washing with methanol. This procedure allows unreacted reagents to be removed and afforded pure oligomers. As a result, the yields of polymers listed in Table range from 54 to 76%.
Table 1. Yields, average molecular weights, and glass transition temperatures of the various polyethers.
The data presented in Table indicate that the molecular weights were relatively low (Mw = 2000–11,000 g mol−1) (values determined by SEC). These values are probably the result of the low solubility of the polyethers obtained in organic solvents. As a consequence, the macromolecular polymer chains leave the organic phase at a certain value of the molecular weight. The low molecular weight can also be explained by the process of transferring bisphenolates to the organic phase.
The effect of temperature on polyether properties was examined. Entries (5, 6, and 7 Table ) show that with increased temperature the values of yield and mass increase and then decrease, in agreement with the general behavior of interfacial polymerization. The best results were achieved in the polycondensation of BPA with the ester n = 6 at 80 °C.
The role of the solvent was also examined. From the data presented in Table (Entries 6, 8, and 9) it follows that the solvent influences the yield and the mass of the polycondensation polymers. The best yield and highest values of mass (for the polycondensation of BPA with the ester n = 6) were obtained with nitrobenzene as organic phase. In fact the polarity of organic solvent influences the effective concentration of catalytic intermediate available for conducting the organic intrinsic reaction so that the product yield and the mass of polymers increase with the increase of solvent polarity.
The effect of the nature of the phase transfer agent was also examined. Different phase transfer catalysts were employed to conduct the present polycondensation reaction. The most active catalyst among those tested is aliquat.
Prepared polyethers using catechol as bisphenol were obtained with low yields compared to those obtained in the case of BPA, their mass was also low, and they are oligomers. These results can be explained by the low reactivity of catechol which has the two OH in ortho position, and is capable of making hydrogen bonding interactions.
3.2. Thermal studies
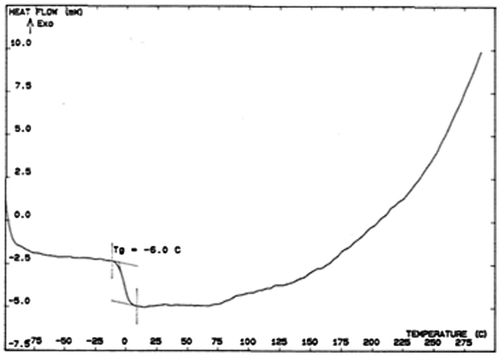
The thermal behavior of the polyethers was evaluated by means of DSC. Two examples are given in Figure (polymer P6c) and Figure (polymer 6 g). They show the usual trace of DSC diagram with a change in the curve characteristic of glass transition. None of the polymers showed clear melting endotherm in the DSC thermograms. This can be attributed to the amorphous nature of the polymers. All polyethers prepared in this study have good stability and the decomposition begins at about 250 °C. (Figures and ).
Figure 5. DSC trace of polymer 6c.
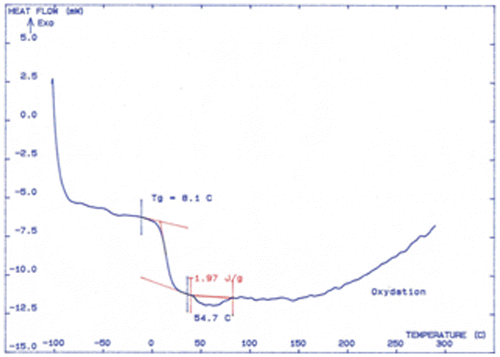
Figure 6. DSC trace of polymer 6 g.
3.3. Hydrolysis of polyethers with esters groups
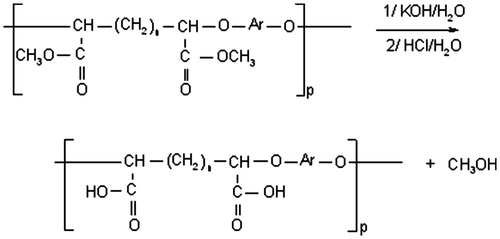
From the obtained polyethers we began to hydrolyze the ester functions in order to have a carboxylic acid function as in the case of type siderophores EDTA.[Citation18] The hydrolysis of these esterspolyethers is in basic medium according to the following reaction (Figure ):
Figure 7. Hydrolysis of polyether ester.
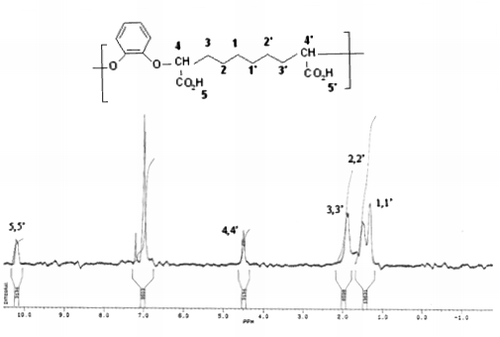
The NMR 1H spectrum of the polymer H1 (Figure ) shows the disappearance of the singlet at 3.6 ppm on proton group (-OCH3) and the appearance of singlet at 10.2 ppm relative to the proton acid which confirms hydrolysis.
Figure 8. 1H NMR spectrum of polymer H1.
4. Conclusions
We have prepared a series of new polyethers by phase transfer catalysis following the polycondensation of diesters dibrominated (EBr2)n with two bisphenols: Catechol and bisphenol A. This is a complex reaction due to the presence of ester functions in the dibromo derivative (EBr2)n, which makes the departure of bromine difficult.
We found that for polyethers with good yields and with high molecular weight, it is best to use aliquat 336 as a catalyst and polar solvents. If we can prepare polyethers with high molecular weight with bisphenol A, we can only find oligomers with catechol, which is less reactive than bisphenol A.
The hydrolysis of the polyethers that contain ester functions gives the polyethers that contain acid functions.
References
- Mathur NK, Williams RE. J. Macromol. Sci. – Rev. Macromol. Chem. 1976;15:117.
- Takemoto K, Ottenbrite RM, Kamashi M. Functional monomers and polymers. New York, NY: Marcel Dekker; 1997.
- Soutif JC, Brosse JC. React. Polym. 1990;12:3.10.1016/0923-1137(90)90058-C
- Zuev VV, Smirnova GS, Nikonorova NA, Borisova TI, Skorokhodov SS. Die Makromolekulare Chemie. 1990;191:2865.10.1002/macp.1990.021911202
- Shin MJ, Kim JG, Shin JS. Int. J. Polym. Mater. 2013;62:140.10.1080/00914037.2011.641641
- Doraswarmy K, Venkata Ramana P. Des. Monomers Polym. 2012;16:56.
- Perumal Bhavani, Sangeetha Dharmalingam. Int. J. Polym. Mater. 2013;62:462.10.1080/00914037.2012.734346
- Jain S, Bajpa AK. Des. Monomers Polym. 2013;16:436.10.1080/15685551.2012.747162
- Sadrolhosseiri AR, Noor ASN, Maarof Moksin MM, Abdi MM, Mohammadi A. Int. J. Poly. Mat. 2013;62:284.
- Bouraoui A, Fathallaha M, Blaive B, Gallo R, Mhenni F. J. Chem. Soc. Perkin Trans. 2. 1990;7:1211.
- Ismail M, Mhenni F, Zinelabidine A, Gallo R. Journal de la Société Chimique de Tunisie. 2002;4:1591.
- Percec V, Schaffer TD. J. Poly. Sci.: Poly. Lett. Ed. 1986;24:439.
- Alazaroaie S, Toader V, Carlescu I, Kazmierski K, Scutaru D, Hurduc N, Simionescu CI. Eur. Polym. J. 2003;39:1333.10.1016/S0014-3057(03)00002-8
- Soga K, Hosoda S, Ikeda S. J. Poly. Sci.: Poly. Chem. Ed. 1979;17:517.
- Nishikubo T. Chemical modification of polymers via phase transfer catalysis. In: Sasson Y, Neumann R, editors. Handbook of phase transfer catalysis. Jerusalem: Springer; 1997. p. 480–509.10.1007/978-94-009-0023-3
- Leung LM, Chik LG. Polymer. 1993;34:5174.10.1016/0032-3861(93)90266-D
- Yang CP, Hsiao SH. Polym. Sci. Polym. Chem. 1990;28:871.10.1002/pola.1990.080280414
- Jacobson L. Plant Physiol. 1951;26:411.10.1104/pp.26.2.411