
Abstract
Novel difunctional monomers are synthesized which are able to dimerize as well as oligomerize via the hetero-Diels–Alder (HDA) mechanism. These monomers are based on 3-aryl-2-cyanothioacrylamides, which are used as functional units for the HDA reaction to form 3,4-dihydro-2H-thiopyrans. Different substituents of the aromatic ring are utilized to shift the equilibrium to the dimeric or oligomeric species. The HDA reaction is reversible and can be influenced by the temperature as well as the concentration. In contrast, the N,N-dimethylthioacrylamide analogues do not dimerize or the dimerization is not reversible. The most promising compound is furthermore investigated in detail by 1H NMR spectroscopy to elucidate the dimerization kinetics. In addition, a difunctional molecule is able to oligomerize reversibly, as confirmed by means of 1H NMR, size exclusion chromatography (SEC), and ESI MS measurements. A respective alcohol is employed as initiator for the ring-opening polymerization of l-lactide resulting in an end-functional polymer that undergoes reversible dimerization as confirmed by SEC measurements.
1. Introduction
In recent years, the research on self-healing materials has developed rapidly due to a strong interest from both sides, fundamental research and application. To achieve intrinsic self-healing abilities of the material, in particular the Diels–Alder (DA) cycloaddition has been applied in many materials, because the original properties of the materials do not change significantly.[Citation1–3] Due to the thermal reversibility of the retro-Diels–Alder (rDA) reaction, it is possible to obtain self-healing polymers with well-defined architectures and properties.[Citation4,5] The DA reaction of furan and maleimide is the commonly used functional system for most reversible self-healing polymers of this type. However, the major drawback of the DA cycloaddition of maleimide and furan is the required high temperature (120 to 180 °C) of the rDA reaction, resulting in self-healing materials that can only be healed when rather drastic conditions are applied.[Citation6,7]
In order to achieve lower rDA temperatures, hetero-Diels–Alder (HDA) systems could represent an interesting alternative for the utilization of reversible crosslinking and post-polymerization functionalization of polymers, respectively. For example, the highly electron-deficient dithioesters that are present as end-groups of polymers obtained via the reversible addition–fragmentation chain transfer polymerization using specially designed chain transfer agents have been utilized for HDA reactions by Barner-Kowollik and coworkers.[Citation8–18] Networks based on tetrafunctional dithioesters and bifunctional cyclopentadiene showed self-healing behavior.[Citation19]
Moreover, Lehn and coworkers investigated highly reversible and dynamic DA systems.[Citation20–22] Various dienes and dienophiles were screened regarding their reversibility of the DA reaction. In particular, functionalized fulvenes have been utilized. These materials, bearing biologically active groups, have been converted with cyano-olefins. Both compounds react rapidly; the DA reaction is reversible in the temperature range from −10 to +50 °C. The variation of these functional units resulted in a new generation of highly dynamic polymers.[Citation20,21] Furthermore, the DA reactions between 9,10-dimethylanthracene and cyano-functionalized dienophiles, e.g., dienes as well as tricyanoethynylethylenes, have been investigated.[Citation22]
Another interesting reaction is the reversible HDA (i.e. dimerization) of 3-aryl-2-cyanothioacrylamides. The first systems were published by Brunskill et al. who reported the thermal dimerization of various 3-aryl-2-cyanothioacrylamides resulting in different derivatives of 3,4-dihydro-2H-thiopyrans.[Citation23,24] 1H NMR and IR data are reported for all 21 thioacrylamides in monomeric and/or dimeric form. Later, the authors investigated the effect of the substituents of the aryl group on the reversible dimerization of 3-aryl-2-cyanothioacrylamides.[Citation24] The equilibrium constants have been determined by 1H NMR spectroscopy. Furthermore, Bloxham and Dell reported the reaction of these 3-aryl-2-cyanothioacrylamides with methyl propiolate and dimethyl acetylenedicarboxylate in 1994 and observed the dimerization of selected derivates.[Citation25]
Due to the reversible dimerization of 3-aryl-2-cyanothioacrylamides, these systems represent interesting candidates for monomers and reversible binding units, respectively. To establish self-healing polymeric or oligomeric systems, the functional moiety should meet two major requirements: a reversible HDA must take place in a ‘user-friendly’ temperature range, and a further functionalization must be possible to incorporate the reversible linkage into polymers.
2. Experimental procedures
2.1. Materials and instrumentation
All chemicals were purchased from Fluka, Aldrich, Acros Organics as well as Alfa Aesar and were used without further purification unless otherwise specified. The solvents were purchased from Biosolve, Aldrich as well as Acros Organics. Benzaldehyde was purified by distillation. Triethylamine was dried over CaCl2. Ethanol was dried by distillation over sodium as well as toluene and THF by distillation over sodium and benzophenone; the solvents were kept under nitrogen using standard Schlenk techniques. l-lactide was purified by recrystallization from dry toluene and dried under vacuum. Sn(Oct)2 was dried by distillation with toluene under reduced pressure. For preparative size exclusion chromatography (SEC), Biobeads SX-1 from BioRad were used.
1D (1H, 13C) nuclear magnetic resonance spectra were recorded on a Bruker AC 400 (400 MHz), Bruker AC 300 (300 MHz), and Bruker AC 250 (250 MHz) at 273, 298, 323, and 343 K. A delay time of 20–30 s was used for the calculations of the monomer to dimer ratio. Chemical shifts are reported in parts per million (ppm, δ scale) relative to the residual signal of the solvent. Coupling constants are given in Hz. The kinetic measurement at room temperature was done manually by daily measurements of the solution in the NMR tube, and the measurements at 50 and 70 °C were annealed and performed in the NMR spectrometer.
MALDI-TOF MS measurements were performed with an Ultraflex III TOF/TOF (Bruker Daltonics, Bremen, Germany) equipped with a smartbeamTM laser and a collision cell. All spectra were measured in the positive reflector or linear mode. The instrument was calibrated prior to each measurement with an external PMMA standard H(CH2CCH3COOCH3)nH + Na+ from Polymer Standards Services (PSS) GmbH (Mainz, Germany) in the required measurement range and trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]-malononitrile (DCTB) (30 mg mL-1 in chloroform) was used as matrix. MS data were processed using the software Flex Analysis and DataExplorer 4.0 from Applied Biosystems (Foster City, CA, USA).
ESI-TOF MS measurements were performed using a micrOTOF (Bruker Daltonics) mass spectrometer, which was equipped with a syringe pump for sample injection and a standard electrospray ion source. The mass spectrometer was operating in the positive ion mode, and the data were processed with the micrOTOF control Version 3.0 and Data Analysis Version 4.0 SP2. The instrument was calibrated by a tunemix solution (m/z 50–3000) from Agilent.
CHN analysis was carried out on a Vario El III (Elementar) elemental analyzer.
SEC measurements were performed using an Agilent 1200 system (degasser, isocratic pump, autosampler, RI detector, DAD) as well as a Shimadzu system (degasser, isocratic pump, autosampler, RI detector) and two PSS GRAM (1000/30 Å, 10 μm particle size) columns in series (eluent: DMA with 2.1 g/L LiCl; flow rate of 1 mL/min at 40 °C) using linear PMMA and PS standards.
The melting points were measured using a Stuart™ melting point apparatus SMP3.
The flash column chromatography was carried out on a Biotage Isolera One System using Biotage SNAP Cartridges KP3Sil and a UV/vis detector.
The ring-opening polymerization (ROP) of l-lactide was performed under nitrogen in a MBraun UNILab glove box workstation.
UV/vis absorption spectra were recorded with a PerkinElmer Lambda 45 UV/vis spectrometer.
2.2. Synthesis
2,2′-(9,9-Didecyl-9H-fluorene-2,7-diyl)bis(1,3,2-dioxaborolane) was synthesized according to Kerszulis et al. with minor adoptions.[Citation26] The aldehydes (1a, 1d, and 1e) as well as 2-cyanoethanethioamide (2b) were purchased from Sigma-Aldrich and were used without further purification, besides benzaldehyde, as described above.
2.2.1. Synthesis of compounds 1b and 1c according to Piñol et al. [Citation27]
2.2.1.1. Synthesis of 4-(11-hydroxyundecyloxy)benzaldehyde (1b)
4-Hydroxybenzaldehyde (2.5 g; 20.47 mmol) was dissolved in 50 mL dry dimethylformamide (DMF). Potassium carbonate (9.5 g; 68.74 mmol) and 11-bromo-1-undecanol (5.1 g; 20.30 mmol) were added, and the resulting mixture was heated for 24 h at 100 °C. Subsequently, the DMF was evaporated and the residue was dissolved in 200 mL chloroform. The organic layer was washed with deionized water (three times 200 mL) and dried over sodium sulfate. The product was obtained by silica gel chromatography (silica, chloroform). Yield: 4.06 g (69%). 1H NMR (CDCl3, 300 MHz): δ = 1.30–1.85 (m, 18H), 3.63 (t, 2H, J = 6.6 Hz), 4.03 (t, 2H, J = 6.6 Hz), 6.98 (d, 2H, J = 8.7 Hz), 7.82 (d, 2H, J = 8.7 Hz), 9.87 (s, 1H) ppm. 13C NMR (CDCl3, 75 MHz): δ = 26.2, 26.4, 29.5, 29.7, 29.8, 29.9, 30.0, 33.2, 63.4, 68.9, 115.2, 130.2, 132.4, 164.7, 191.3 ppm.
2.2.1.2. Synthesis of 3-(11-hydroxyundecyloxy)benzaldehyde (1c)
3-Hydroxybenzaldehyde (2.0 g; 16.38 mmol) was dissolved in 50 mL dry DMF. Potassium carbonate (8.7 g; 62.95 mmol) and 11-bromo-1-undecanol (4.1 g; 16.32 mmol) were added, and the resulting mixture was heated for 24 h at 100 °C. Subsequently, the solution was filtered, the DMF was evaporated, and the residue was dissolved in 200 mL dichloromethane. The organic layer was washed with deionized water (three times 200 mL) and dried over magnesium sulfate. The product was obtained by flash column chromatography (silica, dichloromethane). Yield: 3.9 g (82%). 1H NMR (CDCl3, 300 MHz): δ = 1.30–1.85 (m, 18H), 3.65 (t, 2H, J = 6.0 Hz), 4.01 (t, 2H, J = 6.0 Hz), 7.1–7.2 (m, 1H), 7.3–7.5 (m, 3H), 9.97 (s, 1H) ppm. 13C NMR (CDCl3, 75 MHz): δ = 25.7, 26.0, 29.1, 29.3, 29.4, 29.5, 32.8, 63.0, 68.3, 112.8, 122.0 123.3, 130.0 137.8, 159.7, 192.2 ppm. (C18H28O3)n (292.2)n: Calcd. C 73.93, H 9.65; Found C 74.27, H 9.77.
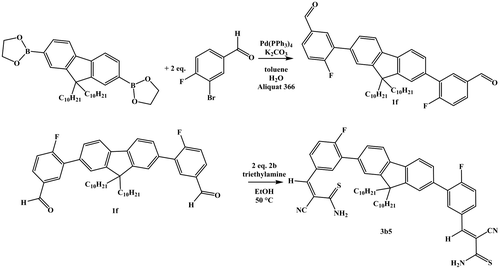
2.2.2. Synthesis of 3,3′-(9,9-didecyl-9H-fluorene-2,7-diyl)bis(4-fluorobenzaldehyde) (1f)
2,2′-(9,9-Didecyl-9H-fluorene-2,7-diyl)bis(1,3,2-dioxaborolane) (100 mg; 0.17 mmol), 3-bromo-4-fluorobenzaldehyde (84 mg; 0.41 mmol) and Pd(0)(PPh3)4 (6 mg; 0.01 mmol) were transferred to a microwave vial under nitrogen atmosphere. Potassium carbonate (113 mg; 0.82 mmol) and 6 drops Aliquat 366 dissolved in 3 mL H2O and 16 mL toluene were added. The reaction mixture was stirred overnight at 120 °C. After cooling to room temperature, the mixture was washed with deionized water and dried over magnesium sulfate. The product was purified by flash column chromatography (silica, dichloromethane/n-hexane 1/1). Yield: 106 mg (90%). 1H NMR (CDCl3, 300 MHz): δ = 0.6–1.4 (m, 38H), 2.0–2.1 (m, 4H), 7.3–7.4 (m 2H), 7.55–7.65 (m, 4H), 7.8–7.95 (m, 4H), 8.05–8.15 (m, 2H) 10.07 (s, 2H) ppm. 13C NMR (CDCl3, 75 MHz): δ = 14.0, 22.6, 23.8, 29.2, 29.5, 29.9, 31.8, 40.2, 55.5, 117.1, 117.4, 120.2, 123.5, 127.9, 130.5, 130.6, 130.8, 132.7, 133.2, 133.3, 140.7, 151.6, 161.9, 165.3, 190.7 ppm. HR-ESI-TOF MS: [C47H56F2O2]Na+ calcd.: m/z = 713.4141; found: m/z = 713.4100; error: 5.7 ppm.
2.2.3. Synthesis of 2-cyano-N,N-dimethylethanethioamide (2a) according to Ransborg et al. [Citation28]
2-Cyano-N,N-dimethylacetamide (1.00 g; 8.92 mmol) was dissolved in 20 mL THF under nitrogen atmosphere. Lawesson’s reagent (1.92 g; 4.73 mmol) was added, and the reaction was stirred for 20 h at room temperature. Subsequently, the solvent was removed under reduced pressure. The product was obtained by flash column chromatography (silica, dichloromethane). Yield: 0.9 g (79%). 1H NMR (CDCl3, 300 MHz): δ = 3.40 (s, 3H), 3.49 (s, 3H), 3.98 (s, 2H) ppm. 13C NMR (CDCl3, 75 MHz): δ = 33.7, 42.2, 44.5, 113.8, 187.6 ppm. (C5H8N2S)n (128.0)n: Calcd. C 46.85, H 6.29, N 21.85, S 25.01; Found C 46.73, H 6.32, N 21.77, S 25.30.
2.2.4. General procedure for the synthesis of compound 3a1–4 and 3b1–5 as adapted from Brunskill et al. [Citation23] Aboutabl et al. [Citation29], and Bloxham et al. [Citation25]
A mixture of 2-cyano-N,N-dimethylethanethioamide (2a) or 2-cyanoethanethioamide (2b), the corresponding aldehyde (1a-1f) (1 or 1.1 eq.) and a base (triethylamine or piperidine) in absolute ethanol was stirred for 30 min to 3 d at 45–50 °C. After absence of precipitation, the residual solvent of the red solution was evaporated under nitrogen stream and further purified.
2.2.5. Synthesis of 2-cyano-N,N-dimethyl-3-phenylprop-2-enethioamide (3a1)
3a1 was synthesized according to the general procedure: 2-Cyano-N,N-dimethylethanethioamide (147 mg; 1.15 mmol), benzaldehyde (1a) (100 mg; 0.94 mmol), triethylamine (0.02 mL; 1.44 mmol), ethanol (2 mL); 1 h at 50 °C. The crude product was further purified by flash column chromatography (silica, dichloromethane/n-hexane 1/1). Yield: 95 mg (47%). 1H NMR (CDCl3, 300 MHz): δ = 3.18 (s, 0.5H), 3.48, 3.50, 3.53 (s, overlapping, 7H), 7.03 (s, 0.2H), 7.35–7.55 (m, 3H), 7.58 (s, 1H), 7.8–7.9 (m, 2H) ppm. (C12H13N2S)n (216.1)n: Calcd. C 66.63, H 5.59, N 12.95, S 14.82; Found C 66.42, H 5.66, N 12.80, S 15.03.
2.2.6. Synthesis of 2-cyano-3-(4-((11-hydroxyundecyl)oxy)phenyl)-N,N-dimethylprop-2-enethioamide (3a2)
3a2 was synthesized according to the general procedure: 2-Cyano-N,N-dimethylethanethioamide (112 mg; 0.87 mmol), 4-(11-hydroxyundecyloxy)benzaldehyde (1b) (204 mg; 0.70 mmol), piperidine (85 μL; 0.84 mmol), ethanol (4 mL); 3 d at 45 °C. The crude product was further purified by flash column chromatography (silica, dichloromethane/ethyl acetate 99/1). Yield: 209 mg (75%). 1H NMR (CDCl3, 300 MHz): δ = 1.1–1.9 (m, 18H), 3.20 (s, 1H), 3.46, 3.50, 3.51 (s, overlapping, 5H), 3.63 (t, 2H, J = 6.0 Hz), 3.9–4.1 (m, 2H), 6.87 (d, 0.5H, J = 9 Hz), 6.94 (d, 2H, J = 9 Hz), 7.34 (d, 0.5H, J = 9 Hz), 7.83 (s, 1H), 7.87 (d, 1.5H, J = 9 Hz) ppm. (C23H34N2O2S)n (402.2)n: Calcd. C 68.62, H 8.51, N 6.96, S 7.96; Found C 68.68, H 8.58, N 6.88, S 7.81.
2.2.7. Synthesis of 2-cyano-3-(3-((11-hydroxyundecyl)oxy)phenyl)-N,N-dimethylprop-2-enethioamide (3a3)
3a3 was synthesized according to the general procedure: 2-Cyano-N,N-dimethylethanethioamide (55 mg; 0.43 mmol), 3-(11-hydroxyundecyloxy)benzaldehyde (1c) (101 mg; 0.34 mmol), piperidine (41 μL; 0.41 mmol), ethanol (2 mL); 3 h at 45 °C. The crude product was further purified by flash column chromatography (silica, dichloromethane/ethyl acetate 99/1). Yield: 114 mg (83%). 1H NMR (CD2Cl2, 300 MHz): δ = 1.2–1.9 (m, 18H), 3.15 (s, 1H), 3.43, 3.46, 3.50 (s, overlapping, 6H), 3.59 (t, 2H, J = 6.0 Hz), 3.85–3.95 (m, 0.5H), 3.95–4.05 (t, 2H, J = 6.0 Hz), 6.9–7.1 (m, 2H), 7.2–7.5 (m, 3H) ppm. (C23H34N2O2S)n (402.2)n: Calcd. C 68.62, H 8.51, N 6.96, S 7.96; Found C 68.84, H 8.53, N 6.65, S 7.60.
2.2.8. Synthesis of 3-(3-bromo-4-fluorophenyl)-2-cyano-N,N-dimethylprop-2-enethioamide (3a4)
3a4 was synthesized according to the general procedure: 2-Cyano-N,N-dimethylethanethioamide (192 mg; 1.50 mmol), 3-bromo-4-fluorobenzaldehyde (1d) (252 mg; 1.24 mmol), piperidine (0.15 mL; 1.50 mmol), ethanol (5 mL); overnight at 50 °C. The crude product was further purified by flash column chromatography (silica, dichloromethane/hexane 1/1). Yield: 145 mg (37%). 1H NMR (CDCl3, 300 MHz): δ = 3.22 (s, 0.5 H), 3.47, 3.51, 3.53 (s, overlapping, 7H), 6.91 (s, 0.2H), 7.1–7.3 (m, 1H), 7.48 (s, 1H), 7.6–7.7 (m, 0.2H), 7.8–7.9 (m, 1H), 8.0–8.1 (m, 1H) ppm. HR-ESI-TOF MS: [C12H10BrFN2S]Na+ calcd.: m/z = 334.9624; found: m/z = 334.9618; error: 1.9 ppm.
2.2.9. Synthesis of 2-cyano-3-phenylprop-2-enethioamide (3b1)
3b1 was synthesized according to the general procedure: 2-Cyanoethanethioamide (100 mg; 1 mmol), benzaldehyde (1a) (117 mg; 1.1 mmol), triethylamine (0.1 mL; 0.72 mmol), ethanol (2 mL); 30 min at 50 °C. The product was obtained by flash column chromatography (silica, dichloromethane/ethyl acetate 99/1). Yield: 30 mg (16%). 1H NMR ((CD3)2CO, 300 MHz): δ = 5.10 (s, 0.1H), 5.28 (s, 0.1H), 6.18 (s, 1H), 7.3–7.4 (m, 1H), 7.5–7.7 (m, 3H), 8.0–8.1 (m, 2H), 8.40 (s, 1H), 8.85 (s, 0.5H), 8.97 (s, 0.1) 9.35 (s, 0.5H) ppm.
2.2.10. Synthesis of 2-cyano-3-(4-hydroxyphenyl)prop-2-enethioamide (3b2)
3b2 was synthesized according to the general procedure: 2-Cyanoethanethioamide (850 mg; 8.49 mmol), 4-hydroxybenzaldehyde (1e) (1 g; 8.49 mmol), triethylamine (0.12 mL; 0.85 mmol), ethanol (20 mL); 30 min at 45 °C. The product was obtained by recrystallization and washing with ethyl acetate. Yield: 173 mg (10%). 1H NMR ((CD3)2CO, 300 MHz): δ = 3.02 (s, 1H), 6.9–7.1 (m, 2H), 7.9–8.1 (m, 2H), 8.41 (s, 1H), 8.61 (s, 1H), 9.48 (s, 1H) ppm.
2.2.11. Synthesis of 2-cyano-3-(3-((11-hydroxyundecyl)oxy)phenyl)prop-2-enethioamide (3b3)
3b3 was synthesized according to the general procedure: 2-Cyanoethanethioamide (173 mg; 1.71 mmol), 3-(11-hydroxyundecyloxy)benzaldehyde (1c) (500 mg; 1.71 mmol), triethylamine (0.01 mL; 0.09 mmol), ethanol (10 mL); 45 min at 45 °C. The product was obtained by flash column chromatography (silica, dichloromethane/ethyl acetate 9/1). Yield: 294 mg (46%). 1H NMR (CD2Cl2, 300 MHz): δ = 1.2–1.9 (m, 18H), 3.59 (t, 2H, J = 6.0 Hz), 4.02 (t, 2H, J = 6.0 Hz), 7.0–7.2 (m, 1H), 7.3–7.5 (m, 1H), 7.5–7.6 (m, 2H), 8.69 (s, 1H) ppm. HR-ESI-TOF MS: [C21H30N2S]Na+ calcd.: m/z = 397.1920; found: m/z = 397.1907; error: 3.3 ppm.
2.2.12. Synthesis of 3-(3-bromo-4-fluorophenyl)-2-cyanoprop-2-enethioamide (3b4)
3b4 was synthesized according to the general procedure: 2-Cyanoethanethioamide (125 mg; 1.25 mmol), 3-bromo-4-fluorobenzaldehyde (1d) (250 mg; 1.25 mmol), triethylamine (0.01 mL; 0.07 mmol), ethanol (5 mL); 1 h at 45 °C. The crude product was washed with boiling dichloromethane. Yield: 75 mg (21%). 1H NMR ((CD3)2CO, 300 MHz, Dimer): δ = 5.10 (s, 1H), 5.36 (s, 1H), 6.37 (s, 2H), 7.2–7.4 (m, 2H), 7.5–7.6 (m, 1H), 7.7–7.8 (m, 2H), 7.9–8.0 (m, 1H), 8.35 (s, 1H), 9.24 (s, 2H) ppm. 1H NMR (CD3)2CO, 300 MHz, Monomer): 7.49 (t, 1H, J = 6.0 Hz), 8.05–8.15 (m, 1H), 8.30–8.33 (m, 1H), 8.33–8.36 (s, 1H) ppm. HR-ESI-TOF MS: [C10H6BrFN2S]Na+ calcd.: m/z = 306.9303; found: m/z = 306.9303; error: 2.7 ppm.
2.2.13. Synthesis of 3-(3-(7-(5-(-3-amino-2-cyano-3-thioxoprop-1-en-1-yl)-2-fluorophenyl)-9,9-didecyl-9H-fluoren-2-yl)-4-fluorophenyl)-2-cyanoprop-2-enethioamide (3b5)
3b5 was synthesized according to the general procedure: 2-Cyanoethanethioamide (30 mg; 0.3 mmol), 3,3′-(9,9-didecyl-9H-fluorene-2,7-diyl)bis(4-fluorobenzaldehyde) (1f) (100 mg; 0.15 mmol), triethylamine (2 μL, 0.01 mmol), ethanol (3 mL); 1 h at 50 °C. The crude product was further purified by flash column chromatography (silica, dichloromethane). Yield: 36 mg (29%). 1H NMR (CD2Cl2, 300 MHz): δ = 0.6–1.3 (m, 38H), 2.0–2.2 (m, 4H), 4.91 (s, 1H, D), 5.20 (0.5H, D), 7.1–7.4 (m, 4H), 7.5–7.95 (m, 10H), 8.0–8.15 (m, 1H), 8.2–8.25 (m, 1H), 8.81 (s, 1H) ppm. HR-ESI-TOF MS: [C53H60F2N4S2]Na+ calcd.: m/z = 877.4120; found: m/z = 877.4065; error: 6.2 ppm.
2.2.14. ROP with compound 3b3 as initiator
l-Lactide (530 mg; 3.68 mmol) was transferred into a microwave vial in a glove box under nitrogen atmosphere. 2-Cyano-3-(3-((11-hydroxyundecyl)oxy)phenyl)prop-2-enethioamide (3b3) (86 mg; 0.23 mmol) dissolved in 3 mL dry toluene and Sn(Oct)2 (50 mg; 0.12 mmol) dissolved in 0.5 mL dry toluene were added to the microwave vial under vigorous stirring. Then, the microwave vial was capped and taken out of the glove box. The reaction mixture was heated at 110 °C in a preheated oil bath for 15 min. The reaction was quenched by addition of 200 μL 1 M HCl solution in methanol. The polymer was purified by precipitation in ice-cold n-hexane and preparative SEC (Biobeads® S-X1, dichloromethane). Yield: 284 mg (46%). Parts of the polymer sample were analyzed by SEC and MALDI-TOF MS to determine the molar mass and molar mass distribution (Mn,MALDI-TOF MS = 1660 g/mol; Mw/Mn = 1.03). 1H NMR (CD2Cl2, 300 MHz): 1.2–1.9 (60H), 3.9–4.0 (1.5H), 4.0–4.2 (1.5H), 4.4–4.4 (1H), 5.0–5.3 ppm (14.5H), 6.7–7.7 (4H), 8.68 (0.2H) ppm.
3. Results and discussion
3.1. Synthesis and characterization of the monofunctional compounds 3a1–4 and 3b1–4
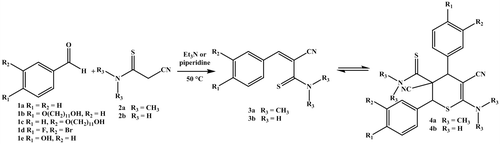
To achieve a highly reversible polymeric structure or end-capped polymer, the first step was to identify suitable compounds, which should be incorporated into a polymeric self-healing material as second step. For this initial screening, eight different compounds 3a1–4 and 3b1–4 were synthesized to investigate the ability to reversibly dimerize via the (retro-)HDA cycloaddition. The synthesis route toward these designed monofunctional compounds with the subsequent dimerization reaction is depicted in Scheme . The first reaction step represents a Knoevenagel condensation of an arylic aldehyde (1) and 2-cyanoethanethioamide (2a or 2b) under basic conditions. This synthetic approach provides access to a series of 3-aryl-2-cyanothioacrylamides with varied substituents on the aromatic ring as well as on the thioamide moieties by combination of different educts. An overview of the, thus, obtained products is provided in Table . For this purpose, five different aldehydes (1) as well as two different 2-cyanoethanethioamides (2a and b) were utilized. Thus, the influence of the substituents could be monitored. Compounds 1b and 1c were synthesized via etherification of p- and m-hydroxybenzaldehyde according to Piñol et al. [Citation27] and compound 2a via thionation of the corresponding aldehyde according to Ransborg et al. [Citation28] The other educts are commercially available. The resulting 3-aryl-2-cyanothioacrylamides (3a and 3b) are potentially able to dimerize via HDA reaction to form 5,6-dihydro-2H-thiopyrans (4a and 4b).
Table 1. Overview of the synthesized monofunctional compounds 3a1–4 and 3b1–4 and their dimerization behavior.
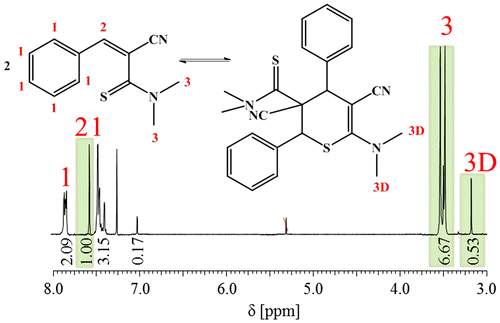
The synthesis and characterization of the 3-aryl-2-cyanothioacrylamides with different substituents and their reversible behavior was also investigated by Brunskill et al. before.[Citation23,24] The thioamide group is a very reactive group, which is favorable with respect to the HDA reaction, but a drawback for the incorporation into a polymeric system, which requires subsequent reaction steps. To overcome the latter restriction, the more stable compounds (3a1–3a4) with the N,N-dimethyl thioamide group were synthesized. 3a1 was synthesized as a model compound to gain insight into the HDA reaction of this less reactive group. 3a1 was obtained with a yield of 47% with triethylamine as base. The characterization via 1H NMR spectroscopy (Figure ) clearly indicates the fact that dimeric species are also formed with the N,N-dimethyl thioamide group. The monomer to dimer ratio of 5.8 to 1 is easily accessible from the integrals of suitable peaks derived from the monomeric (peaks 2 and 3 in Figure ) and the dimeric (peak 3D in Figure ) species.
Figure 1. 1H NMR spectrum of 3a1 and the dimeric species in CDCl3.
3a2 was synthesized to introduce a linker into the system that would be broadly applicable for a number of subsequent reactions, e.g., for post-polymerization modifications. Also here, the 1H NMR spectrum shows clearly a monomer to dimer ratio of 2.3 to 1 (Figure S1). Despite these very promising results, it was not possible to trigger any reversible back reaction to the monomeric species, neither by increasing the temperature up to 70 °C nor by addition of a base such as pyridine or piperidine. An explanation could be the formation of a thermodynamically stable dimeric form due to stereoisomeric rearrangements, which do not dissociate back to the monomeric form. Also for the compounds 3a3 and 3a4 with another linker position and a p-fluoro substitution, which should decrease the electron density within the system, no significant reversibility of the HDA reaction could be achieved. Thus, the dimethyl thioamides seem not to be suitable for the development of self-healing materials in general due to the missing reversibility of the HDA reaction.
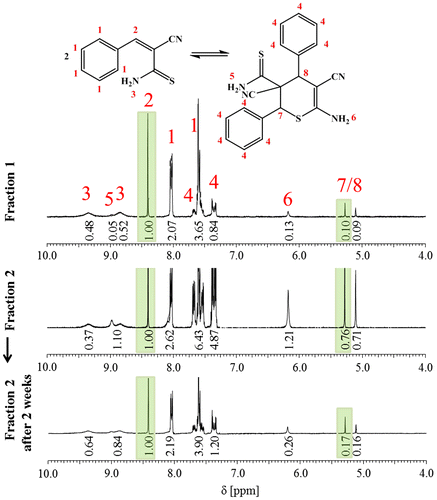
As a consequence, the more labile 3-aryl-2-cyanothioacrylamides without substituents on the amide nitrogen atom were in the focus of further studies. In order to investigate the dimerization and reversibility of the cycloaddition of an already established and working system by studies using 1H NMR spectroscopy, 3b1 was synthesized as a model compound as described by Brunskill et al. [Citation23,24] Figure displays the 1H NMR spectra of two fractions that were isolated via column chromatography directly after the synthesis. The integration of characteristic peaks in the spectra that are originating from monomer (Peak 2 in Figure ) and dimer (Peaks 7 and 8 in Figure ) immediately provides access to the relative amounts of both species that are present. The first fraction revealed a monomer to dimer ratio of 10 to 1. In contrast, the second fraction showed a monomer to dimer ratio of 1.4 to 1. Upon storage of the second fraction at room temperature for two weeks, the amount of dimer is significantly increased ([M]:[D] = 6.4 to 1). Even when the sample is stored at 0 °C for 2 d, the reversibility via HDA reaction can be observed ([M]:[D] = 4.3 to 1). This shows that the ratio could easily be tuned by slight changes of the temperature.
Figure 2. 1H NMR spectra of fraction 1, fraction 2, and fraction 2 after two weeks in solution of 3b1 in (CD3)2CO.
However, to incorporate this very promising HDA system into a polymeric self-healing material, functional moieties that enable a coupling to, e.g., monomers have to be present. Thus, the hydroxyl-functional 3b2 was synthesized. This compound was also synthesized by Aboutabl et al. before.[Citation29] Unfortunately, 3b2 could not be functionalized further via esterification due to the high reactivity of the thioamide group. To overcome this drawback and to enable a preparation of functional polymers containing the cyanothioamide moiety, 3b3 with a long linker and a hydroxyl group was synthesized using triethylamine as base with a yield of 46%. The 1H NMR spectrum showed a monomer to dimer ratio of 24 to 1 directly after synthesis and purification. After storage at 0 °C overnight, the ratio decreased to 4.9 to 1, which clearly indicates the reversibility of the HDA reaction. The application of 3b3 as initiator for the ROP of l-lactide will be discussed below.
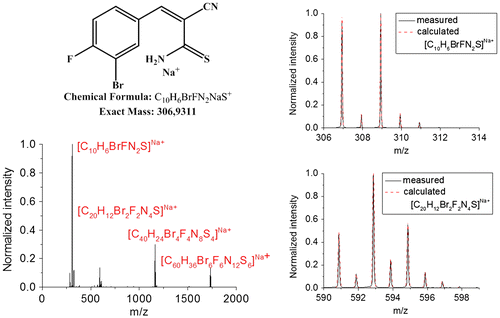
The synthesis of 3b4 was achieved with triethylamine as base. It was selected due to the best dimerization behavior of the substances with the p-fluoro substitution studied by Brunskilll et al. [Citation23,24] After purification only the dimeric species could be isolated, as determined by 1H NMR spectroscopy. In contrast, the ESI-TOF MS spectrum (Figure ) showed the presence of the monomeric as well as dimeric species, but also tetrameric and hexameric species could be assigned, as confirmed by the comparison with the corresponding calculated and measured isotopic patterns (Figures and S2). A possible reason might be the formation of aggregates of dimers that are not separated from each other during the ESI-TOF MS measurement.[Citation30,31] This assumption is supported by the fact that no peaks arising from tri- or pentamers could be observed. Since the m-bromo substitution represents an excellent opportunity to incorporate the 3-aryl-2-cyanothioacrylamide moiety into a polymeric network by cross-coupling reactions, detailed kinetic studies of the dissociation and dimerization of this most promising compound are described in the following.
Figure 3. ESI-TOF mass spectrum of 3b4 with peak assignment and exemplary overlays of the measured and calculated isotopic patterns (monomeric and dimeric species).
3.2. Equilibration studies of 3b4 at different temperatures
For future applications of the functional moiety as key structure in a self-healing polymer, preliminary knowledge about the equilibrium is highly advantageous. For this purpose, detailed kinetic studies were performed via 1H NMR spectroscopy. The retro-HDA reaction was studied at three different temperatures (room temperature, 50, and 70 °C). The equilibration of the corresponding HDA reaction was investigated using a sample that had been thermally pretreated at 70 °C (Figure ).
Figure 4. Equilibration study of 3b4. Data for the rHDA reaction obtained from (CD3)2CO solutions at 25 °C, data for the HDA reaction at 25 °C obtained after annealing at 70 °C in DMSO-d6 (left) and rHDA reaction at 50 °C in (CD3)2CO (right).
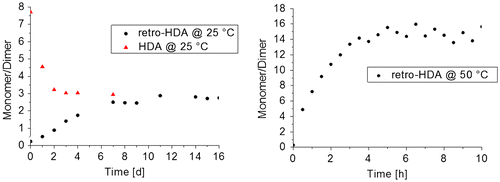
Figure 5. SEC traces of compound 3b5 after multiple concentration steps in CH2Cl2.
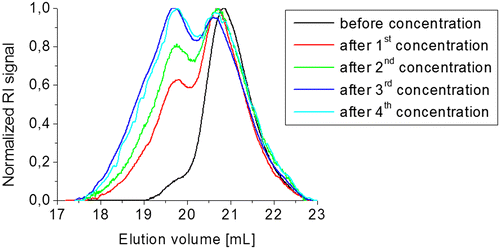
Figure 6. Study of the equilibrium of compound 3b5 at different concentrations in CH2Cl2, (a) directly after dissolving, (b) after 1 d in solution, (c) after 2 d in solution and (d) after 4 d in solution.
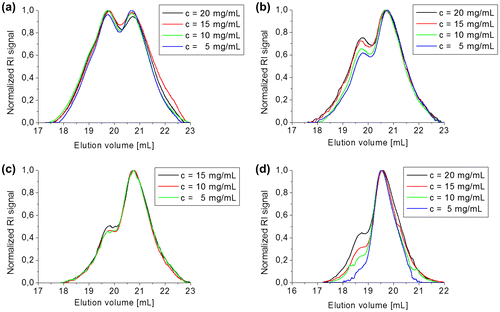
Figure 7. 1H NMR spectrum of 3b5 in CDCl3. For clarity, only the protons and the corresponding peaks that were used for the calculation of the average DP are labelled.
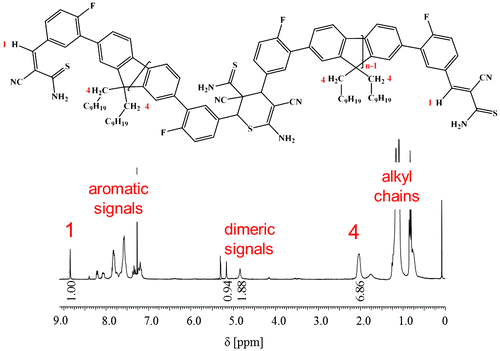
Figure 8. SEC traces of 3b5 after 0, 3, and 6 d in solution (c = 30 mg/mL).
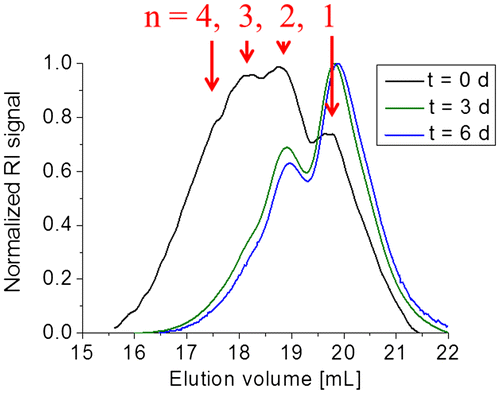
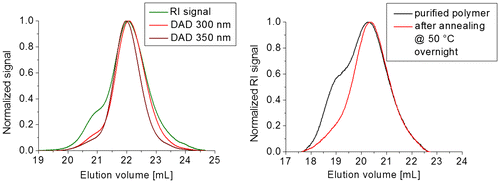
Figure 9. 3D contour plot from the SEC measurement of 3b5 with DAD. The corresponding RID signal is plotted on the right-hand side for comparison.
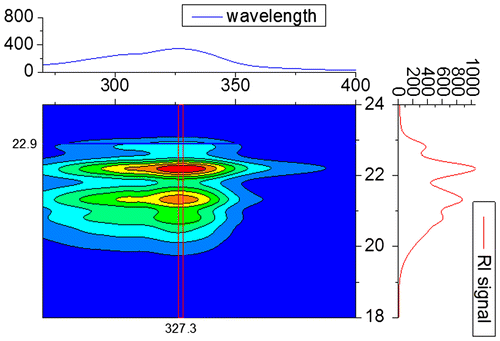
Figure 10. Characterization of P1 via mass spectrometry. Full MALDI-TOF mass spectrum (left) and overlay of calculated and measured isotopic patterns from ESI-TOF MS analysis (right).
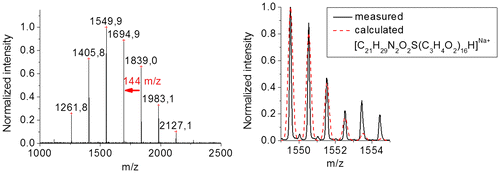
Figure 11. SEC measurement of P1 with RID and DAD (at 300 and 350 nm, the complete contour plot including other is depicted in the supporting information) (left) and SEC traces of P1 before and after annealing at 50 °C (right).
Scheme 1. Schematic representation of the synthesis of the 3-aryl-2-cyanothioacryl-N,N-dimethylamides (3a) and 3-aryl-2-cyanothioacrylamides (3b) as well as the dimerization via HDA reaction.
For the studies of the retro-HDA reaction at room temperature and at 50 °C, acetone was used as solvent, but the studies that required a thermal pretreatment at 70 °C were performed in DMSO due to the higher boiling point. At first, the kinetic study of the equilibration of compound 3b4 at 25 °C in acetone was studied (Figure , left). Two selected 1H NMR spectra are depicted in Figure S3. The ratio of the two species was determined by the utilization of the signals at 5.36 ppm (monomer) and the signals of the aromatic protons at 8.09–9.14 ppm (dimer). At the beginning, only the dimeric species were present; and after 16 days, a ratio of monomer to dimer of around 3 to 1 was detected. The equilibrium was reached after 8 days. A fast increase of the monomer content within the first 8 days was observed. Afterward, only slight changes could be monitored, which are most likely caused by fluctuations due to temperature differences. Furthermore, the influence of the temperature was studied. For this purpose, a kinetic study at 50 °C in acetone was performed, which is depicted in Figure (right). Two selected 1H NMR spectra are shown in Figure S4. Also here, the complete dimeric species was utilized as starting point. Already after 15 min, the monomer to dimer ratio was shifted to 4 to 1. The equilibrium was reached after around 5 h at a monomer to dimer ratio of 15 to 1. In order to further increase the temperature, the solvent was changed to DMSO, which has a higher boiling point. At a temperature of 70 °C, the equilibrium was completely shifted to the monomeric species after 30 min (Figure S5), thereby providing an excellent opportunity to study the equilibration of the corresponding HDA reaction at room temperature. The kinetics of the HDA reaction (Figure , left) showed that the equilibrium was reached after 3 days at room temperature; a monomer to dimer ratio of around 3 to 1 was determined. This shows that the solvent does not influence the composition of the mixture in equilibrium at room temperature and that the HDA reaction is indeed completely reversible.
The fact that the equilibrium is shifted more toward the side of the monomeric species with increasing temperature is typical for DA cycloaddition reactions. Hence, a potential network with crosslinks that are based on the 3b4 functionality would be completely opened already at 70 °C, while the complete reversibility of this particular HDA reaction should provide the possibility to reform the network. Therefore, the incorporation of 3b4 into a polymeric system represented the next important step.
3.3. Synthesis and characterization of 3b5 and the oligomeric species
To incorporate this very promising moiety with a thioamide group and the p-fluoro substitution into a polymer, the difunctional compound 3b5 was synthesized. Since 3b5 contains two cyanothioacrylamide functionalities, it should represent an AA-type monomer that can undergo a reversible polyaddition via ‘dimerization’. Therefore, the fluorene diboronic ester was converted to compound 1f via Suzuki cross-coupling with the corresponding p-fluoro aldehyde with a yield of 90%. The following Knoevenagel reaction toward the difunctional thioacrylamide (3b5) was achieved with a yield of 29% (Scheme ).
Scheme 2. Schematic representation of the synthesis of compound 1f via suzuki cross-coupling and subsequent Knoevenagel reaction to yield 3b5.
The oligomerization of 3b5 already proceeds during the purification of the product. Consequently, analytical techniques that are typical for polymer characterization were applied. 3b5 was further fractionated on a preparative size exclusion Biobeads® S-X1 column. Accordingly, the fractions revealed different Mn values directly after fractionation, as determined by SEC. However, when all fractions were subsequently kept in solution for 5 days, repeated SEC measurements revealed the same molar mass in all fractions. The molar mass of the oligomers tends toward an equilibrium value of 1850 g/mol, which indicates the reversibility of the HDA reaction. Furthermore, we observed that the molar mass could be influenced by the evaporation of the solvent (i.e., the concentration of the monomer). Therefore, different methods were tested (Figure S6) among whose slow evaporation at room temperature was the most effective method in order to increase the molar mass. We also investigated the molar masses of 3b5 after dissolving and evaporating multiple times (Figure ). Also in this case, the Mn value could be increased after several concentration steps. This shows that the dimerization or rather the oligomerization is faster than the retro-HDA reaction toward lower molar mass species.
In a further study (Figure ), the dependency of the molar mass on the concentration in solution was tested. The initial concentration of 3b5 barely influenced the reaction time toward lower molar mass species. Nevertheless, after 4 days in solution, the amount of the larger molar species is only slightly higher at higher concentration. Therefore, the higher molar mass species could only be reached by slow evaporation of the solution.
To further investigate the degree of polymerization (DP) of the higher molar mass species in detail, simultaneous SEC and 1H NMR measurement of the same species were performed. Figure displays the 1H NMR spectrum of 3b5. As can be seen from the schematic representation of the structure of the oligomer, its end-groups will be based on unreacted cyanothioacrylamide moieties making it possible to calculate the average DP. A suitable 1H NMR signal results from the vinylic proton of these end-groups (peak 1 in Figure ). In combination with the signal derived from the methylene protons of the fluorene moieties, a DP of 3.4 (peak 4 in Figure ) could be estimated.
Due to the rather high molar mass of each repeating unit (M = 854 g/mol), the different oligomers could be somewhat resolved by SEC (Figure ) with the dimer being the most abundant species after dissolving the polymer. Upon storage of 3b5 for several days in solution, the DP of the oligomers decreases even further, as confirmed by SEC and 1H NMR measurements. The DP of the oligomers tends toward an equilibrium value 2.
Additional SEC measurements with a diode array detector (DAD) were performed. The corresponding refractive index detector (RID) signal is plotted as reference in the 3D contour plot obtained from the SEC-DAD in the Figures and S7. The intense absorption at ca. 330 nm corresponds to the conjugated oligomer (fluorene flanked by two phenyl moieties). Additionally, the absorption at higher wavelength is caused by the thioacrylamide moiety. Consequently, this absorption is more pronounced for the monomer and dimer, respectively, than for the higher oligomers (only the end-group).
To investigate, if the reversibility of the HDA reaction could be exploited for an application as self-healing material, investigations in bulk are required. In order to obtain preliminary information about an appropriate temperature range, melting point measurements were performed, revealing a Tm around 100 °C. Indeed, the SEC curves of the thermally untreated material show a higher molar mass in comparison with samples that were heated to 100 °C (Figure S8). This demonstrated that the retro-HDA reaction (which results in lower molar masses of the oligomers) indeed takes place in bulk as well.
We also studied the effect of different Lewis acids onto the oligomeric system 3b5 due to the positive influence in other HDA reactions.[Citation32–35] Therefore BF3 etherate as well as ZnCl2 were utilized as Lewis acids with a amount of 0.5 eq per functional unit thioamide. Addition of BF3 resulted immediately in a brown precipitation and further degradation. ZnCl2 induced a degradation of the monomer and reduced the amount of the dimeric species in the solution, as is evidenced by SEC measurement (Figure S9).
To conclude, only oligomeric species could be obtained in solution using the bis functional monomer 3b5 alone. However, due to the highly reversible oligomerization, it was possible to increase the DP by multiple concentration steps. To increase the molar masses of the cyanothioacrylamide-based oligomers while maintaining the high reversibility, the respective functional moiety was therefore combined with a polymer chain that would not be prone to undergo dissociation.
3.4. End-functionalization of polylactide
To introduce the thioamide functionality as end-group to a polymer, the hydroxyl-functional 3b3 was applied as an initiator for the ROP of l-lactide. The resulting polymer could potentially dimerize reversibly.
The ROP was performed under standard conditions using tin(II) bis-(2-ethylhexanoate) as catalyst (Scheme ) with an [M]/[I] of 15 to obtain a short PLA (P1) that would enable a straightforward end-group determination.[Citation36] Despite the unsubstituted thioamide functionality, which could possibly prove problematic during the ROP synthesis of a polyester, PLA with the desired end-group could be obtained. Subsequent to purification, polymer P1 was characterized via SEC, 1H NMR, MALDI, and ESI-TOF MS measurements (Table ). The MALDI-TOF mass spectrum of P1 (Figure ) revealed a single distribution with a m/z difference of 144 between two neighboring peaks, which corresponds to the mass of one lactide monomer. This distribution can be assigned to PLA chains with the monomeric form of the HDA end-group, which are ionized by a sodium cation, as can be seen from the overlay of the measured and theoretical pattern. The high end-group fidelity is also confirmed by the 1H NMR spectrum of P1 (in Figure S10) that clearly shows the characteristic peaks resulting from the four aromatic protons of the initiator between 6.8 and 7.6 ppm. The molar mass of 1680 g/mol, which was estimated using the integrals of these signals and the methine proton signal of the PLA at 5.2 ppm, is in very good agreement to the Mn value from MALDI-TOF MS. Interestingly, a closer inspection of the 1H NMR signal at 8.7 ppm corresponding to the vinyl group proton, already indicates the existence of PLA chains that are coupled via the HDA linkage.
Scheme 3. Schematic representation of the ROP of l-lactide using 3b3 as initiator.
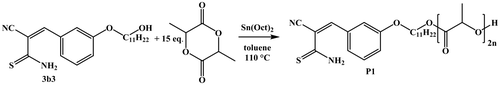
Table 2. Characterization data of the end-functional PLA (P1).
Since a HDA reaction between two end-functional PLA chains directly results in polymer chains with higher molar masses, the SEC trace of P1 (Figure , left) reveals a strong shoulder at lower elution volume, which is in agreement with the considerations based on the 1H NMR spectrum. Due to the UV/vis absorption of the initiator 3b3 until wavelengths λ around 450 nm (Figure S11, left) additional SEC measurements with a DAD (Figure S11, right) were performed. The fact that the polymer can be detected at higher λ once more confirms the covalent attachment of the functional end-group. However, the shoulder arising from HDA-coupled chains is detected with the RID, but can only be seen with the DAD at lower wavelengths. This clearly indicates that the shoulder at higher molar mass corresponds to the dimer. Since the HDA reaction changes the chromophore (compare Scheme ), the UV/Vis absorption properties will strongly be influenced as well.
With respect to a further exploitation of the polymer, the reversibility of the HDA reaction is of major importance and can simply be investigated by SEC in case of P1. As depicted in Figure (right), the high molar mass shoulder, which is present in the purified polymer and originates from the HDA-coupled PLA chains, is not present anymore when P1 is annealed at 50 °C overnight. This demonstrates that the reversibility of the dimerization caused by the used initiator 3b3 is retained when the functional moiety is incorporated in the PLA.
4. Conclusion
Different 3-aryl-2-cyano-N,N-dimethylthioacrylamides (3a1–4) and 3-aryl-2-cyanothioacrylamides (3b1–5) could be synthesized via Knoevenagel reaction. All compounds were characterized via 1H NMR spectroscopy and ESI-TOF MS. The compounds 3a1–4 showed the presence of a dimeric species – the product of the HDA reaction. However, none of these systems was reversible. In contrast, the HDA reaction of 3b1–5 indicated a highly reversible behavior triggered by temperature or concentration. The most promising compound was the p-fluoro-substituted 3-aryl-2-cyanothioacrylamide (3b4), which was consequently selected for kinetic studies of the equilibration. Moreover, this compound was further converted via Suzuki cross-coupling and Knoevenagel reaction to a difunctional species (3b5). The oligomerization and dissociation, which were investigated by SEC measurements as well as 1H NMR spectroscopy, were highly reversible. The molar mass could be adjusted by concentration of the monomer; therefore, the DP could be increased by multiple concentration steps. Nevertheless, the DP was less dependent on the concentration of the solution. Furthermore, 3b3 could be used as initiator for the ROP of l-lactide. The resulting end-functional polyester revealed reversible dimerization. Overall, the dimerization of the 3-aryl-2-cyanothioacrylamides was highly reversible under the investigated conditions (temperature, solvents, concentration,…). On the one hand, this high reversibility is promising for an application in self-healing materials, but on the other hand, the high extent might result in materials with insufficient mechanical properties due to a low cross-link density. The corresponding investigations will be in the focus of our future research.
Supplementary material
The supplementary material for this paper is available online at http://dx.doi.10.1080/15685551.2015.1058007.
Supporting_information_Designed_Monomers_Polymers-2.pdf
Download PDF (467.5 KB)Acknowledgements
Stefan Bode is grateful to the Fond der Chemische Industrie (VCI) for a generous PhD fellowship. Christine Weber acknowledges the Carl Zeiss foundation. Furthermore, Sarah Crotty and Dr. Wolfgang Günther are gratefully acknowledged.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Goussé C, Gandini A, Hodge P. Application of the Diels−Alder reaction to polymers bearing furan moieties. 2. Diels−Alder and retro-Diels−Alder reactions involving furan rings in some styrene copolymers. Macromolecules. 1998;31:314–321.10.1021/ma9710141
- Murphy EB, Bolanos E, Schaffner-Hamann C, Wudl F, Nutt SR, Auad ML. Synthesis and characterization of a single-component thermally remendable polymer network: Staudinger and Stille revisited. Macromolecules. 2008;41:5203–5209.10.1021/ma800432g
- Peterson AM, Jensen RE, Palmese GR. Room-temperature healing of a thermosetting polymer using the Diels−Alder reaction. ACS Appl. Mater. Interfaces. 2010;2:1141–1149.10.1021/am9009378
- Bergman SD, Wudl F. Mendable polymers. J. Mater. Chem. 2007;18:41–62.
- Hager MD, Greil P, Leyens C, van der Zwaag S, Schubert US. Self-healing materials. Adv. Mater. 2010;22:5424–5430.10.1002/adma.201003036
- Chen X, Dam MA, Ono K, et al. A thermally re-mendable cross-linked polymeric material. Science. 2002;295:1698–1702.10.1126/science.1065879
- Chen X, Wudl F, Mal AK, Shen H, Nutt SR. New thermally remendable highly cross-linked polymeric materials. Macromolecules. 2003;36:1802–1807.10.1021/ma0210675
- Sinnwell S, Inglis AJ, Davis TP, Stenzel MH, Barner-Kowollik C. An atom-efficient conjugation approach to well-defined block copolymers using RAFT chemistry and hetero Diels-Alder cycloaddition. Chem. Commun. 2008;2052–2054.10.1039/b718180a
- Inglis AJ, Nebhani L, Altintas O, Schmidt FG, Barner-Kowollik C. Rapid bonding/debonding on demand: reversibly cross-linked functional polymers via Diels−Alder chemistry. Macromolecules. 2010;43:5515–5520.10.1021/ma100945b
- Paulöhrl T, Inglis AJ, Barner-Kowollik C. Reversible Diels-Alder chemistry as a modular polymeric color switch. Adv. Mater. 2010;22:2788–2791.10.1002/adma.201000361
- Espinosa E, Glassner M, Boisson C, Barner-Kowollik C, D'Agosto F. Synthesis of cyclopentadienyl capped polyethylene and subsequent block copolymer formation via hetero Diels-Alder (HDA) Chemistry. Macromol. Rapid Commun. 2011;32:1447–1453.10.1002/marc.v32.18
- Brandt J, Guimard NK, Barner-Kowollik C, Schmidt FG, Lederer A. Temperature-dependent size exclusion chromatography for the in situ investigation of dynamic bonding/debonding reactions. Anal. Bioanal. Chem. 2013;405:8981–8993.10.1007/s00216-013-7203-8
- Guimard NK, Ho J, Brandt J, et al. Harnessing entropy to direct the bonding/debonding of polymer systems based on reversible chemistry. Chem. Sci. 2013;4:2752–2759.10.1039/c3sc50642h
- Oehlenschlaeger KK, Guimard NK, Brandt J, et al. Fast and catalyst-free hetero-Diels–Alder chemistry for on demand cyclable bonding/debonding materials. Polym. Chem. 2013;4:4348–4355.10.1039/c3py00476g
- Pahnke K, Brandt JGryn’ova G, et al. Entropy driven chain effects on ligation chemistry. Chem. Sci. 2015;6:1061–1074.
- Inglis AJ, Sinnwell S, Stenzel MH, Barner-Kowollik C. Ultrafast click conjugation of macromolecular building blocks at ambient temperature. Angew. Chem. Int. Ed. Engl. 2009;48:2411–2414.10.1002/anie.200805993
- Inglis AJ, Stenzel MH, Barner-Kowollik C. Ultra-fast RAFT-HDA click conjugation: an efficient route to high molecular weight block copolymers. Macromol. Rapid Commun. 2009;30:1792–1798.10.1002/marc.v30:21
- Langer M, Brandt J, Lederer A, Goldmann AS, Schacher FH, Barner-Kowollik C. Amphiphilic block copolymers featuring a reversible hetero Diels-Alder linkage. Polym. Chem. 2014;5:5330–5338.10.1039/C4PY00644E
- Oehlenschlaeger KK, Mueller JO, Brandt J, et al. Adaptable hetero Diels–Alder networks for fast self-healing under mild conditions. Adv. Mater. 2014;26:3561–3566.10.1002/adma.v26.21
- Boul PJ, Reutenauer P, Lehn J. Reversible Diels−Alder reactions for the generation of dynamic combinatorial libraries. Org. Lett. 2005;7:15–18.10.1021/ol048065k
- Reutenauer P, Buhler E, Boul PJ, Candau SJ, Lehn J. Room temperature dynamic polymers based on Diels-Alder chemistry. Chem. Eur. J. 2009;15:1893–1900.10.1002/chem.v15:8
- Reutenauer P, Boul PJ, Lehn J. Dynamic Diels-Alder reactions of 9,10-dimethylanthracene: reversible adduct formation, dynamic exchange processes and thermal fluorescence modulation. Eur. J. Org. Chem. 2009;2009:1691–1697.10.1002/ejoc.v2009:11
- Brunskill JSA, De A, Ewing DF. Dimerisation of 3-aryl-2-cyanothioacrylamides. A [2s+ 4s] cyclo-addition to give substituted 3,4-dihydro-2H-thiopyrans. J. Chem. Soc., Perkin Trans. 1. 1978:629–633.10.1039/p19780000629
- Brunskill JSA, De A, Ewing DF. Substituent effects on a reversible cycloaddition reaction. J. Chem. Soc., Perkin Trans. 2. 1980:4.
- Bloxham J, Dell CP. Reaction of 3-aryl-2-cyanothioacrylamides with electron-deficient alkynes: synthesis of 4-aryl-4H-thiopyrans. J. Chem. Soc., Perkin Trans. 1. 1994:989–993.10.1039/p19940000989
- Kerszulis JA, Amb CM, Dyer AL, Reynolds JR. Follow the yellow brick road: structural optimization of vibrant yellow-to-transmissive electrochromic conjugated polymers. Macromolecules. 2014;47:5462–5469.10.1021/ma501080u
- Piñol R, Lub J, García MP, et al. Synthesis, properties, and polymerization of new liquid crystalline monomers for highly ordered guest−host systems. Chem. Mater. 2008;20:6076–6086.
- Ransborg LK, Albrecht Ł, Weise CF, Bak JR, Jørgensen KA. Optically active thiophenes via an organocatalytic one-pot methodology. Org. Lett. 2012;14:724–727.10.1021/ol203237r
- Aboutabl MA, Abdel Aziz MA, Magd El Din AA, Elwy HA, Fahmy HM. Electrochemical reduction of some 3-aryl-2-cyanothioacrylamide derivatives at the DME. Monatsh. Chem. 1991;122:765–770.10.1007/BF00815916
- Di Marco VB, Bombi GG. Electrospray mass spectrometry (ESI-MS) in the study of metal-ligand solution equilibria. Mass. Spectrom. Rev. 2006;25:347–379.10.1002/(ISSN)1098-2787
- Schug K, McNair HM. Adduct formation in electrospray ionization mass spectrometry. J. Chromatogr. A. 2003;985:531–539.10.1016/S0021-9673(02)01732-6
- Motoki S, Saito T, Karakasa T, et al. Lewis acid-promoted hetero Diels-Alder reaction of α, β-unsaturated thioketones. J. Chem. Soc., Perkin Trans. 1. 1992:2943–2948.10.1039/p19920002943
- Saito T, Fujii H, Hayashibe S, Matsushita T, Kato H, Kobayashi K. Uncatalysed (thermal) and Lewis acid-promoted asymmetric hetero-Diels–Alder reaction of 1-thiabuta-1,3-dienes (thiochalcones) with di-(–)-menthyl fumarate. Configuration determination by x-ray crystallographic analysis of (2s,3r,4r)-(+)-2,3-bis[(–)-menthoxycarbonyl]-4,6-diphenyl-3,4-dihydro-2h-thiopyran and conversion of cycloadducts into optically pure diols. J. Chem. Soc., Perkin Trans. 1. 1996:1897–1903.10.1039/p19960001897
- Bastin R, Albadri H, Gaumont A, Gulea M. Pyridinedithioesters as heterodienophiles: application to the synthesis of Aprikalim. Org. Lett. 2006;8:1033–1036.10.1021/ol052942k
- Zhou J, Guimard NK, Inglis AJ, et al. Thermally reversible Diels–Alder-based polymerization: an experimental and theoretical assessment. Polym. Chem. 2012;3:628–639.
- Kryuchkov MA, Detrembleur C, Jérôme R, Prud’homme RE, Bazuin CG. Synthesis and thermal properties of linear amphiphilic diblock copolymers of L-lactide and 2-dimethylaminoethyl methacrylate. Macromolecules. 2011;44:5209–5217.