
Abstract
Hexamethylene diisocyanate (HMDI) based polyurethane-urea-amide (PUUA) is synthesized using diamide chain extenders and dihydroxy polystyrene. The rigid segment structure is HMDI-6X6-HMDI. The repeating molecular weight of the soft segment is varied from 2678 to 9202 g mol−1 using HMDI as a chain extender. The inherent viscosity of the polymer is 1.3–3.9 dL g−1. Tm of PUUA varies between 239 and 265 °C. The crystallinity of the amide segments is up to 70%. The polymers show single-stage decomposition temperature around 415 °C. WAXS data confirm the semi-crystalline nature of the polymer. The solvent resistivity of the polymer in chloroform is high due to high crystallinity of the hard segment.
1. Introduction
Linear polyurethane elastomers consist of alternating mobile polystyrene segments, and rigid urethane or urea segments are classified as thermoplastic polyurethanes (TPU’s). Due to incompatibility of the alternating hard and soft segments (SS), microphase separation easily occurs. The rigid segment’s phase separates into hard domains which is dispersed in a soft matrix. The hard phase acts as a thermoreversible physical crosslinks which allows melt processing. Phase separation either occurs by liquid–liquid demixing or crystallization of the rigid segments. Liquid–liquid demixing leads to aggregation of rigid segments, resulting in spherical hard domains that hardly contribute to the reinforcement of the polymer. Liquid–liquid demixing is favoured by the presence of long rigid segments and can be avoided using short uniform rigid segments.[Citation1] Most commercial TPU’s have a random distribution of rigid segments resulting in a complex morphology and a low degree of crystallinity, normally lower than 20%.[Citation2] When the rigid segments are dissolved in the soft phase, the Tg of the soft phase is increased and broadened, resulting in a poor low-temperature flexibility.
To induce phase separation by crystallization, monodispersed crystallizable segments can be used.[Citation3–5] It is difficult to synthesize polyurethanes with monodisperse rigid segments via melt polymerization and also processing of these polymers without losing the uniformity of rigid segments is difficult. The dispersity of the rigid segments depends on the polymerization procedure. For having a uniform hard segment (HS), the polymerization temperature should be low, the prepolymer should not contain any free diisocyanates and the extender should be uniform in length. Free diisocyanates in the prepolymer can react with an extender and form longer blocks. Another reason to use prepolymers with low diisocyanate contents is to avoid the toxicity of the formed polymer.
Amide groups are thermally more stable than urethane groups and also amide-interchange reactions during high-temperature melt polymerization hardly occur.[Citation6] Exchanging some of the urethane groups in TPU’s with amide groups decreases the urethane content and thereby might increase the thermal stability. In several studies, polyurethanes with uniform amide extenders were reported.[Citation1,4,5] Versteegen [Citation5] prepared block copoly(ether urea)s with amine-terminated prepolymer as flexible segments via protective group strategy and novel isocyanate chemistry. Segmented block copolymers with uniform rigid segments were synthesized by Van der Schuur et al. [Citation6] These polymers with monodisperse rigid segments had higher strength and toughness compared to analogous polymers with polydisperse rigid segments. Heijkants [Citation7] studied polyurethanes based on poly(ε-caprolactone) end capped with 1,4-butane diisocyanate and 1,4-butanediol as extender. These studies show that polyurethanes with monodisperse rigid segments based on aliphatic isocyanate end cappers can have high moduli. Van der Schuur et al. made poly(ether-urethane-urea-amide)’s (PEUUA) from poly(propylene oxide) end capped with 2,4-toluene diisocyanate (2,4-TDI) and uniform amide extenders (e.g. 6T6-diamine diamide).[Citation1]
Polystyrenes are high-Tg (100 °C) materials extensively used in electrical, electronics, medical, home application, packaging industry, etc. The amorphous polystyrene, like other amorphous polymers, possesses poor solvent resistance and environmental stress cracking. To improve the solvent resistance of amorphous polymers, they are often blended with a high melting semi-crystalline material at a higher quantity (>40 wt.%).[Citation8,9] Also, amorphous polystyrene has been modified by reacting with diisocyanates to form semi-crystalline polyurethane. In this way, a semi-crystalline copolymer is obtained and it is found to have both amorphous soft-segment and crystalline hard-segment phases having different chemical structures. However, the low-crystallization window of high-Tg materials decreases the crystallization rate of the HS and crystallinity.[Citation10]
Polyurethanes are thermally instable and can have a complex morphology, which complicates the study on the effect of hydrogen bonding on polymer properties. Segmented polyamides are thermally stable and have a two-phase morphology and can therefore be studied in a simplified manner. Therefore, the aim of this study is to synthesize the segmented polyurethane-urea-amide (PUUA) based on dihydroxy polystyrene end capped with 1,6-hexamethylene diisocyanate (HMDI) and diamine-diamide extenders like 6A6 or 6T6. The properties of the PUUA copolymers derived from 6A6 and 6T6 are compared.
2. Experimental
2.1. Materials
1,6-Hexamethylenediisocyanate, phenol and 1,1,2,2-tetrachloroethane, catalyst dibutyltin dilaurate (DBTDL), deuterated triflouroaceticacid (TFA) and hexaflouroisopropanol (HFIP) are purchased from Aldrich Chemicals Co. Tetrahydrofuran, toluene, methanol and dimethylacetamide (DMAc) are purchased from S.D fine chemicals and used after distillation. The functionalized hydroxy-terminated polystyrene (OH-PSt-OH) is synthesized using the known route.[Citation11] The 6T6 and 6A6 diamine-diamide are synthesized using the known route.[Citation12]
2.2. General procedure for the synthesis of block copolymer (Scheme )
Segmented block copolymers are synthesized using various molar ratios of soft and hard segments, with two different chain extenders (6A6 or 6T6). The synthetic routes of the block copolymer are presented in Scheme . The mole ratios taken for the synthesis are presented in Table . Preparation of the sample code A1 is given here as an example. A similar procedure is followed for the preparation of other copolymers. The reaction is carried out in a 250-ml stainless steel vessel with a nitrogen inlet and a mechanical stirrer. The reactor is charged with 6 g of PSt2000 (0.003 mol) and 1.1 g (0.006 mol) of HMDI dissolved in 20 ml of DMAc. To this, 2–3 drops of DBTDL are added as catalyst. The stirred reaction mixture is heated to 80 °C for 3 h. To this, 1.03 g (0.003 mol) of 6A6 dissolved in 20 ml of DMAC is added and temperature was subsequently raised to 150 °C over a time period of 2 h. Thus, the polymer is formed. The copolymer is dried overnight in vacuum at 70 °C for lateral use. In the solid state, the polymer is transparent with a slight cream hue. Yield: 9.1 g (88%). 1H NMR (δ, ppm): 6.6–7.4 (b, aromatic proton), 5.0 (b,>NH), 3.3–3.6 (m, –CH2- connected to urea and amide group), 3.0 (b, −CH2- connected to urethane group) 1.2–1.8 (b, aliphatic proton).
Table 1. Mole fraction in feed, repeating length and the inherent viscosities of the polymers.
2.3. Fouriertransform infrared spectroscopy
Infrared spectra were recorded using Alpha Bruker FT-IR with a resolution of 2 cm−1. Samples were prepared by adding a droplet of polymer solution (HFIP (1 g L−1)) on a pressed KBr pellet and the measurements were carried out at room temperature. Temperature-dependent FT-IR measurements are recorded on an Alpha Bruker instrument connected with alpha-T accessory and recorded between 30 and 120 °C under a nitrogen flow. The samples investigated for the purpose of this work, are prepared from the 5 wt.% solution of PUUA in HFIP. The films are prepared by pouring the solution onto a potassium bromide pellet and by evaporating the solvent. These conditions allow the polymer films to have a stable morphology with desirable properties.[Citation13]
The degree of crystallinity of the rigid segments in the polymers could be estimated with the following equations:(1)
The heights (h) of the amorphous and crystalline urethane peaks were related by the factor ‘‘C’’ with a value of 2.4.[Citation14]
2.4. Inherent viscosity measurements
The inherent viscosity of the copolymers at a concentration of 0.1 g dL−1 in a 1:1 (molar ratio) mixture of phenol/1,1,2,2-tetrachloroethane is determined at 25 °C using a capillary Ubbelohde.(2)
where, t = Elution time of the polymer solution, t0 = Elution time of the solvent and c = Concentration of the polymer solution (g dL−1).
2.5. Wide angle X-ray scattering (WAXS)
Wide-angle 2D-X-ray scattering is measured using the Xeuss WAXS instrument. Cu-K-alpha is the X-ray source. Sample to theta distance is 221.75 mm and silver behenate is used for as distances calibration standard. 2D-X-ray scattering data is integrated using Fit-2D software.
2.6. NMR
1H NMR spectra of the copolymers are run on a Bruker FT-NMR spectrophotometer operating at 320 MHz at room temperature using deuterated TFA as a solvent and tetramethylsilane (TMS) as an internal reference.
2.7. Differential scanning calorimetry
DSC spectra are recorded on a Perkin Elmer DSC 7 apparatus equipped with a PE 7770 computer and TAS-7 software. About 10–15 mg of dried copolymer sample is heated at a rate of 20°C min−1 for recording DSC spectra. The second heating and first cooling curve are used to evaluate the Tm and Tc of HS, respectively.
2.8. Thermogravimetric analysis
Thermal stability of the polymers is done using a thermogravimetric analysis (TGA) method using Dupont 951 thermo gravimetric analyzer. The sample weight is 8–10 mg. The work is performed from 30 to 600 °C at a heating rate of 10 °C min−1 in a nitrogen atmosphere with a gas flow rate of 100 ml min−1.
2.9. Swelling ratio of polymer
The equilibrium water absorption (WA) is measured on pieces of injection-moulded polymer bars. The samples are placed in desiccators with a layer of demineralized water for four weeks at room temperature. WA is defined as the weight gain of the polymer according to Equation (3).(3)
where m is the weight of the dry sample and m0 the weight of the sample after conditioning to equilibrium.
2.10. Solvent resistivity
Injection-moulded samples of dimension 10 × 10 × 2 mm are used for solvent resistance measurements. Previously weighed samples are dipped in 50 ml of organic solvents taken in a flask and shaken for 60 min. After that, the solvent is decanted and the flask is dried for 24 h at 70 °C. It is weighed again and from the weight loss, the amount of polymer sample dissolved in the solvents is calculated.
(4)
where, mo is the weight of dry the substance (mg) before solvent treatment, m is the weight of the substance (mg) after solvent treatment.
3. Results and discussion
3.1. Synthesis of poly(urethane-urea-amide)
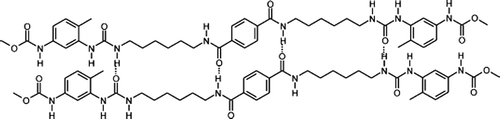
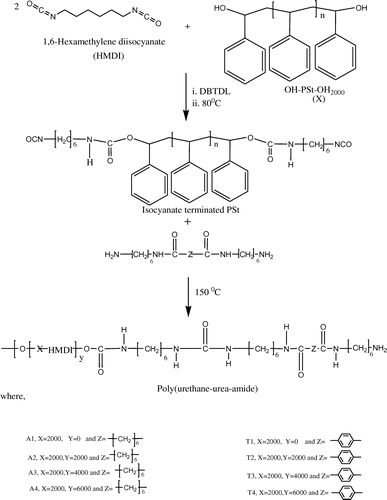
PUUAs are synthesized by a two-step polyaddition reaction. In the first step, a prepolymer is synthesized using one mole of hydroxyl terminated polystyrene (soft segment (SS)) and two moles of diisocyanate (HMDI).Reaction of the hydroxy group with the isocyanate resulted in an isocyanate endcapped prepolymer containing urethane linkage. In the second step, the isocyanate endcapped polymer is reacted with different diamine–diamide chain extender (6A6 (A series) or 6T6 (T series)), thereby increasing the molecular weight. The chemical structure of PUUAs is shown in Scheme . The synthesized compounds are characterized by FT-IR and NMR techniques. The NMR data confirm the presence of styrene, urea and urethane linkage. The presence of multiple peaks around 3.3–3.6 ppm confirms that the polymer contains a CH2 group, which is connected to the amide group. This proves that the HS, i.e. diamide (6A6) segment is effectively incorporated in the polymer chain, thereby it increases the molecular weight. Though the NMR is not conclusive for the formation of the desired multiblock copolymer (it only gives the constituents present in the polymer chain), nevertheless it is qualitatively shown some idea about the polymer structure.
3.2. Inherent viscosity measurements
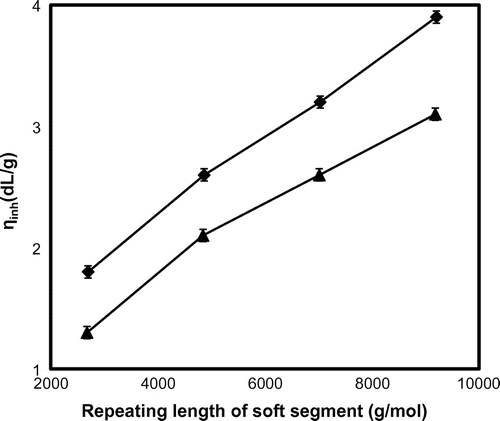
Inherent viscosity of the copolymer is determined in phenol/1,1,2,2-tetrachloroethane (1:1 molar mixture) to get an idea about the molecular weight. The OH-PSt-OH shows the inherent viscosity of 0.1 dL g−1. The inherent viscosity of A and T series polymer is about 1.3–3.1 and 1.8–3.9 dL g−1, respectively. It is expected that the increase in molecular weight of polymer is evident from the increase in inherent viscosity. In both the series, the inherent viscosity is linearly increasing with increasing the repeating unit of the SS (Table ). The correlation between the repeating molecular weight of the SS to that of inherent viscosity is presented in Figure .
Figure 1. Effect of soft segment length on the molecular weight of the polymer.
3.3. FT-IR
Infrared spectroscopy is a common tool to study the hydrogen bonding in segmented block polyurethanes and is also used for the PUUA copolymers. In HMDI-based PUUA copolymers, three groups which are present that can form H-bonds are urethane, urea and amide. The absorbance bands of these carbonyls groups in free and in hydrogen bonded state are reported in the literature.[Citation15–17] Urea carbonyl can form two types of assemblies namely: bidendate (strongest) and monodendate as shown in Figure .
Figure 2. Possible hydrogen bonding interactions of urea, urethane and amide groups.
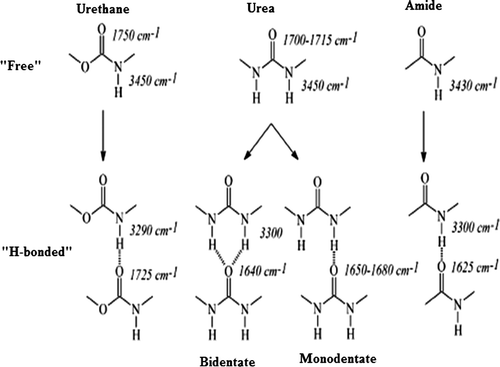
Figure shows the room temperature FT-IR spectra of the polymers A1 representing (-(T-PSt)y-PSt-T-6A6-T)-n and T1 representing (-(T-PSt)-PSt-T-6T6-T)-n; and the calculated crystallinity of the amide group is presented in the Table . Prominent peaks observed in the FT-IR spectrum are 3310–3329, 1669–1690 and 1624–1635 correspond to crystalline N–H, urea and amide carbonyls, respectively.
Figure 3. FT-IR spectrum of two copolymers at room temperature: ♦, A1; ▲, T1.
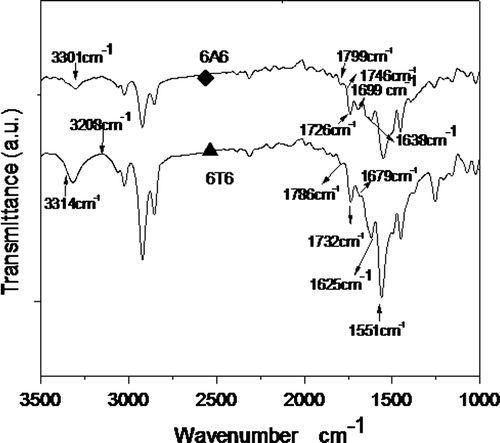
Table 2. FT-IR data of the multiblock copolymer at room temperature.
The crystallinity of the segmented block copolymers containing crystallizable (by hydrogen bonding) HS can be studied by FT-IR spectroscopy technique. These measurements can be done by looking at the absorption peaks of the carbonyl groups present in the HS. The absorbance band of the crystalline amide carbonyl groups is located at 1625 cm−1 and that of the amorphous amide carbonyl is at 1679 cm−1. For calculating crystallinity in copolymer, the peak height of 1625 cm−1 and the peak height of 1679 cm−1 are taken to represent the crystalline and amorphous states of the amide carbonyl, respectively. The synthesized multiblock copolymers have a strong crystalline amide peak at 1625 cm−1 and a weak peak corresponding to the amorphous amide carbonyl at 1679 cm−1. This indicates that most of the HS are in the crystalline state. But, as the HS contents decrease in the copolymer, the crystallinity decreases suggesting a less close packing of HS in the lengthy SS. The crystallinity of the terephthalic acid-based HS (70%) is more than that of adipic acid (60%) suggesting that the aromatic group facilitates a better close packing of crystals than the aliphatic counterpart (Table ).[Citation18]
3.4. Temperature dependent FT-IR
The effect of temperature on the structure and properties of PUUA is investigated by in situ FT-IR (Figure ). To investigate the changes in the hydrogen bonds distribution resulting from the thermal treatment, the spectra of PUUs are recorded as a function of increasing and the subsequent decreasing temperature, and analysed with respect to the peak shape, intensity and the position shift. The N–H stretching region has been shown to be inadequate to characterize the hydrogen bonding due to the presence of urethane, urea and amide groups. The carbonyl region, on the other hand, has been widely used to characterize the hydrogen-bonding state of the polymer. Figure shows the FT-IR spectrum of the polymer T1 at different temperatures as a representative of the studied series. This graph shows that the crystallinity of the synthesized polymer changes slightly upon increasing the temperature form 40 to 120 °C. This polymer holds its high crystallinity even at a temperature of 120 °C. This remarkable stability arises due to the uniform HS, which is present in the polymer chain.
Figure 4. Temperature dependent FTIR spectrum of T1 copolymer at different temperatures.
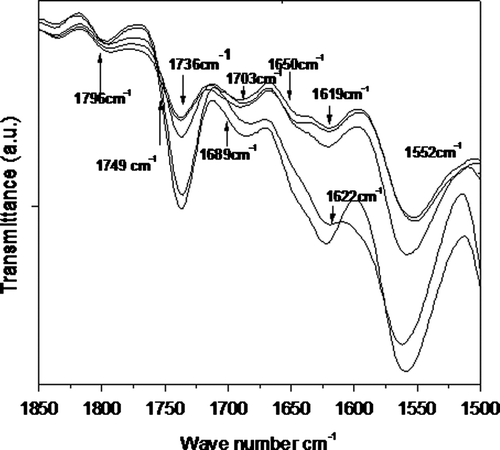
A similar type of spectrum is obtained for other samples in the series. Upon heating, the intensity of crystalline amide peak decreases and intensity of amorphous amide peak increases as shown in Figure . Using this difference in intensity between crystalline and amorphous amide peaks, we calculate the percentage of crystalline region. The crystallinity of the HS decreases with increasing temperature. This effect is more pronounced in the A1 sample than in the T1. This is due to the percentage of the crystalline region is high in the T series compared to A series, which is presented in Table . Also the Tm values of the T-series polymers are higher than those of A series. This high crystallinity is due to effective phase separation facilitated by the terephthalic group present in the HS. The possible interaction between the HS is presented in Figures and .
Figure 5. Schematic diagram of the hydrogen bonding interaction in 6A6 HS.
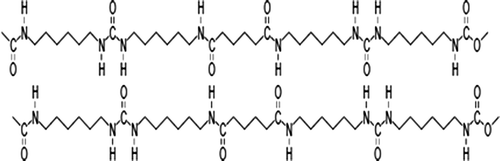
Figure 6. Schematic diagram of the hydrogen bonding interaction in 6T6 HS.
3.5. WAXS
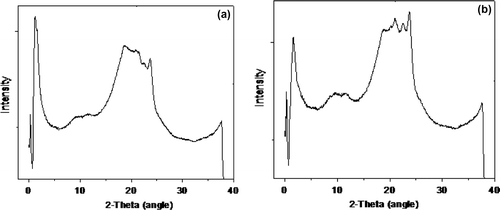
The WAXS graph of the A1 and T1 sample is presented in Figure , and the A1 graph shows two prominent peaks at 1.3 and 23.55 two thetas corresponding to two different planes and the length of the crystalline planes are 6.39 and 17.82 A°, respectively . Also, the two peaks at 2 theta values of 1.3 and 23.55° correspond to (0 0 1) and (1 0 0) planes. These data are shown in Tables and .[Citation19] WAXS pattern of T1 shows four peaks observed at 1.8, 20.99, 22.60 and 23.73 two θ corresponding to four planes and the length of the crystalline planes are 7.9, 11.5, 12.9 and 17.8 A0, respectively, and corresponding to (0 0 1) and (1 0 0) are observed. This clearly suggests that sample T1 formed a better crystalline structure than A1. The 6T6 present in T1 encourages greater ordering of the HS resulting in high crystallinity in the soft matrix. The fractional crystallinity present in the WAXS curve is calculated utilizing the software program from the internet.[Citation20] The crystallinity calculated using the software is around 60% at 30 °C and this high value is in accordance with the crystallinity calculated by FT-IR measurements. Further, it is concluded that the HS are completely phase separated as a crystal, and therefore we get sharper peaks in the WAXS experiments.[Citation21]
Figure 7. WAXS pattern of the sample at room temperature: (a), A1; (b), T1.
3.6. DSC
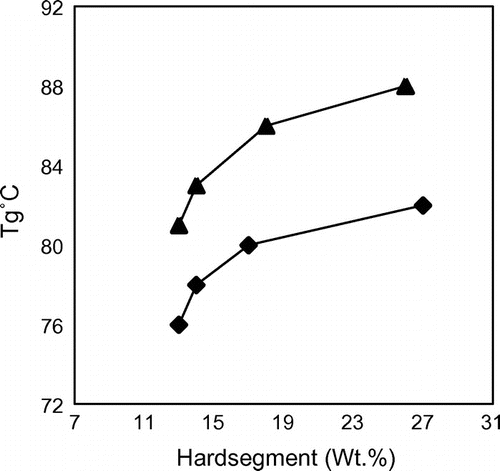
The DSC thermograms of the segmented polyurethane are analysed to gather maximum information concerning their structure. The DSC spectra of A1 and T1 are given in Figure and the results are summarized in Table . The heating curve of the polymer sample shows the Tg of SS and Tm of HS. The cooling curve of A1 and T1 shows crystallinity exotherm. The Tg value of the polymers T1–T4 starts at 88 °C and ends at 81 °C. Similarly, the Tg value starts at 82 °C and ends at 76 °C for A1–A4 series. This can be due to the increase in rigidity provided by the HS which decreases chain flexibility and consequently increases Tg. The Tg obtained for the T-series polymers having similar HS content are higher than those obtained from A series. This is due to the presence of rigid and bulky aromatic ring in 6T6 which facilitates better crystallinity for the HS, and therefore lesser HS in the soft matrix. The effect of HS content to that of the Tg is presented in Figure .
Figure 8. DSC heating curve and cooling curve of two polymer: ♦, A1; ▲, T1.
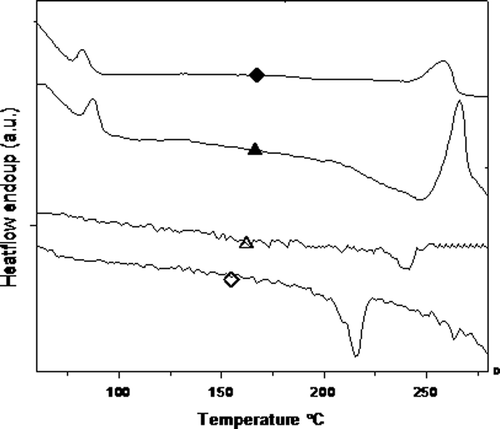
Table 3. DSC, TGA, swelling ratio and solubility data of the polymer at room temperature.
Figure 9. Effect of HS concentration on the Tg of the soft phase: ♦, 6A6 series; ▲, 6T6 series.
The Tm value of the polymers T1–T4 is found to lie between 265 and 243 °C. It shows that the melting temperature decreases with decreasing HS content. Similarly, in case of A1–A4, it is found to lie between 258 and 239 °C. It clearly indicates that the melting temperature decreases with decreasing HS content. The Tm value is higher (having similar HS content) in the case of 6T6 than 6A6; it means that the percentage of crystalline region is more in 6T6 than in 6A6. The values of Tm–Tc indicate the crystallization rate, which is given in the Table . The Tm–Tc value of T1 is low compared to A1 series due to the comparatively large crystallization window of the former than latter. T1 copolymer has low undercooling values and this in turn suggests that the monodisperse rigid segment crystallizes very fast.
3.7. TGA
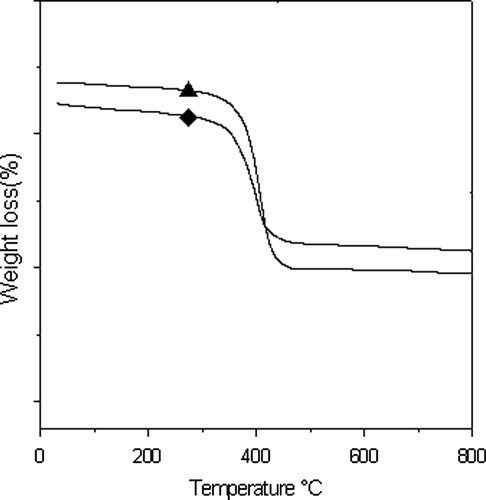
TGA curves of the polymer samples A1 and T1 are given in Figure and the data are presented in Table . The initial decomposition temperature is taken as the onset point. The decomposition temperature values of copolymer based on 6A6 are found between 414 and 416 °C. Similarly, the decomposition temperature for 6T6-based polymer is found between 416 and 418 °C. All the materials show the single-stage decomposing temperature around 415 °C, which clearly shows a high thermal stability of the synthesized block copolymers. The thermal stability of the block copolymers are slightly higher than the pure PSt (412 °C) which are employed in the synthesis process and can be due to the incorporation of the HS in to the PSt SS.
Figure 10. TGA thermograms of the two polymer: ♦, A1; ▲, T1.
Scheme 1. The reaction pathway for the synthesis of semi-crystalline copolymer.
3.8. Swelling ratio of polymer
The effect of HS content on WA is presented in Table . PUUA are comes under semi-crystalline polymer and therefore physical crosslinks present between the polymer chains. The density of physical crosslinking depends on the HS content. The physical crosslinking point opposes the water molecule to penetrate through the soft matrix. Therefore, on increasing the HS, the swelling ratio decreases. The water molecules can penetrate into the polymer matrix in two ways: one is free water that can freely transfer from holes to polymer chains and the other one is bound water that can interact with functional groups of material. Extensive moisture absorption leads to the increase in bound water to interact with functional groups of PUUA.[Citation22] Water molecules, which penetrate the inner structure of PU matrix will act as a plasticizer to generate hydrogen bonding between N–H and C=O groups to permit polymer chains to move freely . The WA value of the A series polymers are higher than those of T series due to low crystallinity and/or high hydrophilic character of A.
Table 4. WAXS data for PUUA based A1 and T1.
3.9. Solvent resistivity
Polystyrenes can be easily attacked by solvents, but the solvent resistance of semi-crystalline polymer can be improved. Solvent resistance behaviour of multiblock copolymers is studied by suspending injection-moulded bars in chloroform at room temperature for 1 h. After drying the samples, the weight loss is determined. The PSt dissolved completely in chloroform within 1 h as shown in Table . The multiblock copolymer has dramatic lower weight losses in chloroform and in other organic solvents. Although the solubility decreases with increasing HS content, the observed high values of solubility are due to the highly polar nature of the urethane–urea linkage present in the polymer chain. Similar to WA behaviour, the solvent resistivity of A-series copolymer is lesser than that of T-series copolymer (Table ).
Table 5. Simple peak indexing for PUUA based A1 and T1.
4. Conclusion
PUUA with different concentrations of HS and different chain extender (6A6 or 6T6) have been prepared and characterized by NMR, FT-IR, WAXS, DSC and TGA. The molecular weight of the polystyrene is increased from 2678 to 9202 g mol−1 using HMDI as a chain extender and thereby changing the HS concentration from 13 to 27 wt.%. The crystallinity of the HS was measured by FT-IR techniques and shows the high value for the T series (up to 95%). The monodisperse hard segments crystallize fast and completely. WAXS measurement confirms the formation of copolymer with excellent crystallinity for the 6T6-based PUU. DSC data of the synthesized segmented copolymer show two transition, Tg of SS and the melting of HS as expected for semi-crystalline materials. Values of Tm are higher in case of 6T6 than in 6A6-based PUUA; it means that the percentage of crystalline region increases in former than latter. The undercooling value of these materials is low, suggesting fast crystallization of HS. The thermal stability of these materials is very high and the decomposition temperature is revolved around 415 °C, which clearly shows a high thermal stability of the synthesized block copolymers. The swelling and the solubility of the polymer (in chloroform) is found to depend on the type of the chain extender and the concentration of the HS.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Van der Schuur M. Poly(propylene oxide) based segmented block copolymers [PhD thesis]. Netherlands: University of Twente; 2004.
- Martin DJ, Meijs GF, Gunatillake PA, McCarthy SJ, Renwick GM. The effect of average soft segment length on morphology and properties of a series of polyurethane elastomers II SAXS-DSC annealing study. J. Appl. Polym. Sci. 1997;64:803–817.10.1002/(ISSN)1097-4628
- Eisenbach CD, Stadler E, Enkelmann V. Synthesis and properties of molecularly uniform oligourethanes with chain-folding units in the center. Macromol. Chem. Phys. 1995;196:833–856.10.1002/macp.1995.021960313
- Heijkants RGJC. Design, synthesis and properties of a degradable polyurethane scaffold for meniscus regeneration. J. Mater. Sci. Mater. Med. 2004;15:423–427.10.1023/B:JMSM.0000021114.39595.1e
- Versteegen RM. Well-defined thermoplastic elastomers [PhD thesis]. Netherlands: University of Eindhoven; 2003.
- Schuur M, Feijen J, Gaymans RJ. Synthesis and characterization of bisester-amide segments of uniform and random length. Polymer. 2005;46:4584–4595.10.1016/j.polymer.2005.02.074
- Heijkants RGJC. Polyurethane scaffolds as meniscus reconstruction materials [PhD thesis]. Netherlands: University of Groningen; 2004.
- Bates FS, Hillmyer MA, Lodge TP, Bates CM, Delaney KT, Fredrickson GH. Multiblock polymers: panacea or pandora's box? Science. 2012;336:434–440.10.1126/science.1215368
- Pêgo AP, Poot AA, Grijpma DW, Feijen J. Physical properties of high molecular weight 1,3-trimethylene carbonate and D, L lactide copolymers. J. Mater. Sci. Mater. Med. 2003;14:767–773.10.1023/A:1025084304766
- Zhang L, Eisenberg A. Multiple morphologies of “crew-cut” aggregates of polystyrene-b-poly(acrylic acid) block copolymers. Science. 1995;268:1728–1731.10.1126/science.268.5218.1728
- Vakees E, Suresh J, Kayalvizhi M, Srinivasan VV, Arun A. Synthesis and characterization of functionalized polystyrene using ATRP with strong base technique. IOSR - J. Appl. Chem. 2014;7:32–38.10.9790/5736
- Krijgsman J, Husken D, Gaymans RJ. Synthesis and characterisation of uniform bisester tetra-amide segments. Polymer. 2003;44:7043–7053.10.1016/S0032-3861(03)00681-5
- Wolińska-Grabczyk A. Relationships between permeation properties of the polyurethane-based pervaporation membranes and their structure studied by a spin probe method. Polymer. 2004;45:4391–4402.10.1016/j.polymer.2004.04.039
- Arun A, Konraad D, Gaymans RJ. The melt rheological behavior of AB, ABA, BAB, and (AB) block copolymers with monodisperse aramide segments. Macromol. Chem. Phys. 2009;210:48–59.
- Kayalvizhi M, Vakees E, Suresh J, Nagarajan S, Arun A. Spacer length controlled highly thermo reversible polyurethane-urea based on polystyrene – synthesis and crystallization studies. Polym. Adv. Technol. 2015;26:160–166.10.1002/pat.v26.2
- Dounis DV, Wilkes GL. Structure property relationships of flexible polyurethane foams. Polymer. 1997;38:2819–2828.10.1016/S0032-3861(97)85620-0
- Kaushiva BD, Wilkes GL. On the issue of urea phase connectivity in formulations based on molded flexible polyurethane foams. Polymer. 2000;41:285–310.
- Biemond GJE, Krijgsman J, Gaymans RJ. Poly(ether amide) segmented block copolymers with adipic acid based tetraamide segments. J. Appl. Polym. Sci. 2007;103:512–518.
- Theivasanthi T, Alagar M. X-ray diffraction studies of copper nano powder. Archives Phys. Res. 2010;1:112–117.
- http://coecs.ou.edu/Brain.P.Grady/saxssoftware.html.
- Suhas DP, Jeong HM, Aminabhavi TM, Raghu AV. Polymer Engineering Science. 2014;54:24–32.10.1002/pen.v54.1
- Yu Y-J. The effect of moisture absorption on the physical properties of polyurethane shape memory polymer foams. Taiwan: National Taiwan University; 2011.