
Abstract
Starting from the key intermediate 1′,2,3,3′,4,4′-hexa-O-benzylsucrose, two bifunctional monomers (1′,2,3,3′,4,4′-hexa-O-benzyl-6,6′-dimethacryloylsucrose and 6,6′-diallyl-1′,2,3,3′,4,4′-hexa-O-benzylsucrose) were synthesized, applying microwave-assisted protocols. Two types of hydrophobic polymer beads were obtained by copolymerization with styrene, in which the bifunctional sucrose monomers acted as cross-linkers. The particles were characterized by degree of cross-linking, Tg (measured by DSC), particles form and size by AFM, and their properties compared.
1. Introduction
The use of biodegradable polymeric micro- and nanoparticles for controlled drug delivery has shown significant therapeutic potential.[Citation1–3] Nanoparticles are defined as colloidal particles of a diameter less than 100 nm, while particles with diameter from 100 nm up to one micrometer are referred to as microparticles.[Citation4,5] They are widely employed in various fields of the life sciences, such as separation technologies, histological studies, clinical diagnostic assays, and drug delivery systems.[Citation6] A very important advantage of naturally derived polymers as drug delivery systems over synthetic analogs is their innate biocompatibility and biodegradability,[Citation7] but due to the complexity of the natural polysaccharides, it is difficult to introduce fine structural modification for specific applications. That is why we aim to combine the advantages of natural compounds and synthetic systems and we have reported the synthesis of various linear, soluble sucrose-containing polymers.[Citation8–10] The biodegradability of polymers with a polyvinyl backbone with pendant saccharide moieties has been reported previously.[Citation11–13] It has also been shown that copolymers with sucrose moieties protected with acetyl or benzyl groups were able to biodegrade, although slower than the corresponding copolymers with unprotected (free hydroxyl) sucrose moieties.[Citation14,15]
The developments in this field have been summarized by Queneau et al. [Citation16], and some notable advances in this area are listed below. Dordick et al. have reported the synthesis of poly(sucrose acrylate) and poly(sucrose adipamide) with the help of an enzyme (Proleather).[Citation17] Spherical sucrose-containing polymer beads with tailored morphology have been obtained and used in solid-phase peptide synthesis.[Citation18] Hou et al. [Citation19] have also prepared polysucrose microspheres by reversed suspension polymerization. A novel viscous surfactant based on polymerized sucrose and using microwave irradiation was reported.[Citation20] In another study, a cross-linked sucrose polymer with dithiothreitol as comonomer was prepared by the thiol-ene photopolymerization technique.[Citation21]
During this work, we aimed to prepare cross-linked beads by suspension polymerization of styrene, in which the bifunctional sucrose monomers acted as cross-linkers. Similar structures with various applications have been reported by other groups,[Citation18,19,22] but in contrast to these reports, our approach was to synthesize chemoselectively structurally well-defined bifunctional sucrose monomers. For this, our key intermediate was 1′,2,3,3′,4,4′-hexa-O-benzylsucrose 1, which was obtained as previously reported.[Citation23,24] Two bifunctional monomers, 1′,2,3,3′,4,4′-hexa-O-benzyl-6,6′-dimethacryloylsucrose 2 and 6,6′-diallyl-1′,2,3,3′,4,4′-hexa-O-benzylsucrose 3 were synthesized and copolymerized with styrene to obtain hydrophobic polymeric microparticles, which were characterized by the degree of cross-linking (Tg) and the particle form and size by AFM.
2. Results and discussion
2.1. Synthesis of sucrose monomers
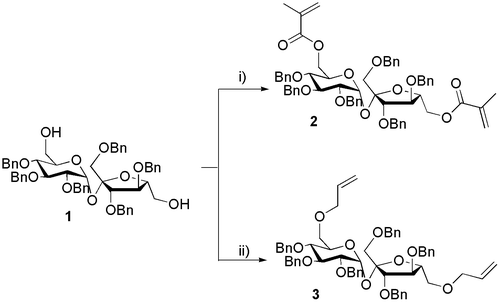
Of the various polymerizable substituent groups reported,[Citation25] methacryloyl ester and allyl ether groups were selected as representatives of the two different classes of compounds. Thus, two different monomers were obtained (Scheme ), using compound 1 as starting material.
Esterification of the hydroxyl groups of 1 was carried out under microwave irradiation in a mixture of dichloromethane and triethylamine (Et3N) with methacrylic anhydride, to afford the expected compound 2 in good yield (65%); and under conventional conditions in order to compare the outcome, which was similar (69%). The 1H-NMR spectra of this compound exhibited two sets of signals, which have been attributed to the two double bonds – two singlets at 6.09 and 6.06 ppm for the α proton and two singlets at 5.56 and 5.46 ppm for the β proton. Two signals were observed at 1.94 and 1.85 ppm corresponding to the two methyl groups.
Conventional etherification involving a primary halide, in our case allyl bromide,[Citation26,27] was performed and optimized under microwave irradiation. Thus, the alcohol 1 was treated with NaH in DMF at 0 °C, followed by addition of allyl bromide to form 3 in 74% yield after microwave irradiation (145 °C, 300 W, 10 min).
2.2. Synthesis of cross-linked polymeric beads
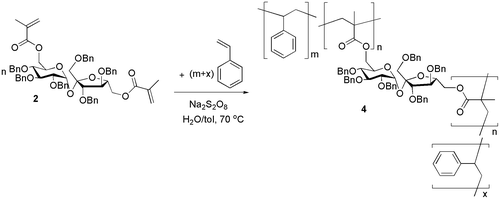
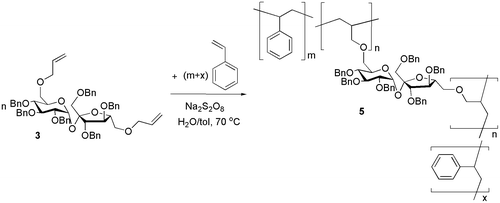
Using the sucrose monomers 2 and 3, two different types of polymeric microparticles were synthesized by free radical dispersion polymerization (Schemes and , Table ). The polymerization conditions, reaction time, and the initial monomers ratio have been optimized as previously described.[Citation9,10] The microparticles reported herein have been obtained with an initial monomer ratio sugar–styrene 1:10, but depending on the application intended, the composition of the copolymer and subsequently the density of the cross-linked network can be varied by modifying the initial feed.
Table 1. Free radical copolymerization of sucrose monomers 2 and 3 with styrene.
Two solutions were prepared: a water solution of the emulsion stabilizer polyvinyl alcohol (PVA) 3% w/v and the radical initiator sodium persulfate, Na2S2O8 (1% with respect to the monomers mixture); and a toluene solution containing the hydrophobic sugar monomer (2 or 3) and styrene in a molar ratio of 1:10. An oil-in-water emulsion was formed by combining the two solutions and dispersing them by vigorous stirring. This emulsion was heated at 70 °C for 24 h with vigorous stirring. After cooling, the heterogeneous mixture was centrifuged to collect the produced microparticles and these were washed with distilled water and then cold methanol to remove any residual monomers, and freeze-dried. An insoluble powder of cross-linked polymeric microparticles 4 (yield 39% based upon the weight of the monomers mixture) and 5 (yield 37% based upon the weight of the monomers mixture) was obtained. All the reported experiments have been triplicated in order to confirm the reproducibility of the methods, and the reported characteristics are the numerical averages of the obtained data.
Direct estimation of the degree of cross-linking was attempted by two different methods [Citation28,29] – by IR spectroscopy and the amount of gel fraction after extraction using CH2Cl2 as a solvent and a Soxhlet extractor. The IR spectra of the solid polymer samples 4 and 5 did not show any signals corresponding to the vinyl groups of the monomers, which led to conclusion that any unreacted monomers had been removed during the work-up.
The degree of cross-linking by the amount of gel fraction can be found as a ratio between the sample mass after extraction and the initial sample mass, in percentages. In our case, four hours extraction in a Soxhlet extractor did not yield any soluble fractions for the two samples, which corresponds to degree of cross-linking 100%. Therefore, to estimate indirectly the constitution of polymer 4, the amount and constitution of the unreacted monomers mixture after isolating the solid particles of 4 were analyzed. By 1H-NMR of this residue, it was estimated to contain 5.3 mol% of sugar monomer 2, and 94.7 mol% styrene. From this data, knowing that the monomers feed ratio was sugar/styrene 1:10 and that the yield of solid polymer was 39%, the monomers units contained in the obtained polymer 4 have been calculated to be 15 mol% sugar and 85 mol% styrene, or sugar/styrene ratio 0.18. Assuming that the sugar and styrene units were randomly distributed in the polymer chains, the length of the linear polymer segments was estimated to be on the average around three styrene units.
Respectively, the soluble fraction recovered from polymer 5 was found to contain 9.7 mol% of sugar monomer 3, and 90.3 mol% styrene. The yield of solid polymer was 37%, which contained 8.1 mol% sugar and 91.9 mol% styrene, or sugar/styrene ratio 0.09. Assuming that the sugar and styrene units were randomly distributed in the polymer chains, the length of the linear polymer segments was estimated to be on the average around six styrene units. This difference can be explained by the different reactivity ratios of the two monomers and the fact that the methacryl group under these conditions polymerizes faster than the allyl group.
2.2.1. Differential scanning calorimetry (DSC)
DSC traces of the two polymer samples can be seen in the ESI, and the polymer’s glass transition temperatures (Tg) are presented in Table . The polymers synthesized were amorphous, having only a glass transition temperature, and neither crystallization nor melting temperatures were observed. The two polymers 4 and 5 exhibited Tg-s with a difference of ten degrees (85.7 and 75.4 °C, respectively), which was in accordance with previously published results for similar polymers.[Citation8,30] For the polymer 4, which had twice the cross-linked density of 5, the observed Tg was approximately 10° higher than for the polymer 5, containing longer linear polymer segments. This result suggested that the presence of a higher percentage of cross-linking in the polymeric matrix promoted an increase in the polymer Tg, which indicated a decrease of the polymer chains flexibility and malleability.
2.2.2. Atomic force microscopy (AFM)
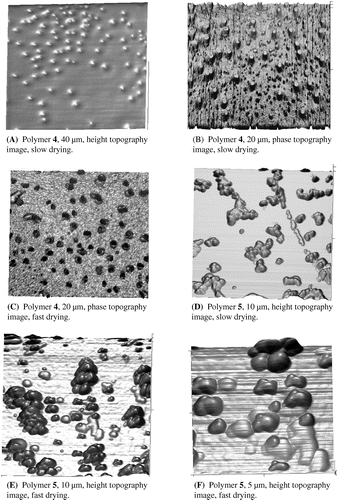
The surface morphology and size distribution of the polymer samples were examined by AFM and representative images are shown in Figure . The films were obtained by suspending the polymeric microparticles in distilled H2O, followed by depositing the dilute dispersion onto freshly cleaved mica and drying. The results were greatly dependent on the conditions used for the preparation of the films such as particles concentration, nature of the solvents used, speed, and temperature of drying, etc. Under the selected conditions, described in the experimental part, it was observed that both polymers 4 and 5 formed uniformly shaped microparticles. The images of the more densely cross-linked polymer 4 (Figure (A)–(C)) show spherical polymeric beads with sizes ranging from 600 to 750 nm when dried slowly and 380 to 420 nm when dried under vacuum. The less densely cross-linked polymer 5 (Figure (D)–(F)) tended to form variously shaped clusters of microparticles, with sizes ranging from 600 to 800 nm depending on the drying method.
Figure 1. AFM images of polymeric particles 4 and 5.
Scheme 1. Synthesis of sucrose-containing monomers from 1′,2,3,3′,4,4′-hexa-O-benzylsucrose 1. (i) (CH2=C(CH3)CO)2O, CH2Cl2, Et3N, DMAP, 35 ºC, 300 W, 10 min, 65% or rt, 4 h, 69%. (ii) CH2=CH–CH2–Br, NaH, DMF, 145 °C, 300 W, 10 min, 74% or rt, 3 h, 79%.
Scheme 2. Synthesis of the cross-linked polymer 4.
Scheme 3. Synthesis of the cross-linked polymer 5.
3. Conclusions
In summary, the synthesis of new sucrose-containing monomers was developed using both conventional and microwave heating, the outcome being focused on the selectivity of the transformations. From the key intermediate 1′,2,3,3′,4,4′-hexa-O-benzylsucrose 1, two bifunctional monomers 1′,2,3,3′,4,4′-hexa-O-benzyl-6,6′-dimethacryloylsucrose 2 and 6,6′-diallyl-1′,2,3,3′,4,4′-hexa-O-benzylsucrose 3 were synthesized using microwave-assisted protocols, which allowed significant reduction of time and energy and then characterized. From these key monomers, the cross-linked copolymers poly(1′,2,3,3′,4,4′-hexa-O-benzyl-6,6′-di-O-methacryloylsucrose)-co-polystyrene 4 and poly(6,6′-di-O-allyl-1′,2,3,3′,4,4′-hexa-O-benzylsucrose)-co-polystyrene 5 were formed and converted to micro-sized hydrophobic particles by dispersion polymerization in aqueous media. These were characterized by the polymers constitution, degree of cross-linking, Tg measured by DSC, and particle form and size by AFM.
4. Experimental section
4.1. General methods
All reagents and solvents were purified before use.[Citation31] The reactions under microwave irradiation were performed in open flasks equipped with temperature control sensor and magnetic stirring using a monomodal microwave reactor MicroSynth Labstation (MileStone, USA). The reaction conditions are expressed as a function of the reaction temperature, which was the controlled parameter, and not the magnetron power. NMR spectra were recorded at 400 MHz in CDCl3 or DMSO-d6, with chemical shift values (δ) in ppm downfield from TMS (0 ppm) or the residual solvent peak of DMSO (2.50 ppm). The signals were assigned with the aid of DEPT, COSY, and HMQC experiments. Optical rotations were measured at 20 °C on an AA-1000 polarimeter (0.5 dm cell) at 589 nm and are expressed in deg cm3 g−1 dm−1. The concentrations (c) are expressed in g 100 cm−3. FTIR spectra were recorded on Perkin–Elmer Spectrum BX apparatus in KBr dispersions or on NaCl cells. DSC measurements were carried out on a Setaram DSC 131 scanning calorimeter equipped with a thermal analysis data system. Samples of 10 mg were placed in aluminum pans and sealed. The probes were heated two times, from −20 to 80 °C at a rate of 10 °C/min and from 25 to 250 °C under nitrogen atmosphere. MALDI-TOF spectra were recorded on Ultraflex III TOF/TOF Bruker equipped with laser-type smartbeam and detecting system fast MCP-Gating. AFM images were acquired on a TT-AFM instrument from AFM Workshop in a vibrating mode. The polymer films were obtained by suspending the particles in distilled H2O, followed by depositing the dilute dispersion onto freshly cleaved mica, then thorough drying in an exicator either at atmospheric pressure overnight or under vacuum for 2 h.
4.2. 1′,2,3,3′,4,4′-Hexa-O-benzyl-6,6′-di-O-methacryloylsucrose 2
To a 0.1 M solution of 1 (500 mg, 0.56 mmol) in anhyd CH2Cl2, Et3N (260 mg, 2.56 mmol) and a catalytic amount of DMAP was added. The mixture was cooled to 0 °C, and then, 0.5 M solution of methacrylic anhydride (210 mg, 0.20 mL, 1.36 mmol) in anhyd CH2Cl2 was added. The reaction mixture was stirred at 0 °C for 10 min, and then placed in the microwave cavity and subjected to microwave irradiation (max 300 W at constant temperature 35 °C) for 10 min (microwave conditions) or stirred at rt for 4 h (conventional conditions). The mixture was diluted with more CH2Cl2 (40 ml) and washed with aq 1.0 N HCl (8 ml), saturated aq NaHCO3, and distilled H2O. The organic layer was dried over anhyd Na2SO4, and the solvent evaporated. Purification by flash column chromatography, eluent hexane- EtOAc, 5:1, yielded 370 mg, 65% (microwave conditions) or 400 mg, 69% (conventional conditions) of 2.
[α]D20 + 53.2 (c = 0.7, CHCl3); no report in the literature. IR: νmax (NaCl): 3029(C–H, stretch, unsaturated), 2923 (C–H, stretch, saturated), 1720 (C = O), 1495, 1453 (C–C, stretch), 1295 (C–H, assym.), 1165 (C–C–O), 1089 (C–O–C, C–O–H, stretch), 1013, 944 (O–C–C, st as, ester, C–O–C, st sym, ether), 736, 697 (Ar) cm−1.
1H NMR (400 MHz, CDCl3) δ: 7.39–7.21 (m, 35H, Ar–H), 6.09 (s, 1H,=CH2a), 6.06 (s, 1H,=CH2a), 5.74 (d, J1,2 = 3.4 Hz, 1H, H-1), 5.56 (s, 1H,=CH2b), 5.46 (s, 1H,=CH2b), 4.97 (d, J = 10.6 Hz, 1H, Ar–CH2), 4.87 (d, J = 10.8 Hz, 1H, Ar–CH2), 4.80 (d, J = 10.6 Hz, 1H, Ar–CH2), 4.69–4.40 (m, 12H, Ar–CH2, H-3′, 4′, 6), 4.36–4.25 (m, 3H, H-1′, 6′), 4.22–4.15 (m, 2H, H-1′, 5), 4.14–4.07 (m, 1H, H-5′), 4.06–3.99 (m, 1H, H-3), 3.96 (d, J = 9.2 Hz, 1H, Ar-CH2), 3.73 (d, J = 10.8 Hz, 1H, Ar-CH2), 3.60–3.51 (m, 2H, Ar-CH2, H-4), 3.47 (dd, J1,2 = 3.7 Hz, J2,3 = 9.6 Hz, 1H, H-2), 1.94 (s, 3H, CH3), 1.85 (s, 3H, CH3).
13C NMR (100 MHz, CDCl3) δ: 167.0, 166.9 (COO–), 138.7, 138.2, 138.1, 137.9, 137.8, 137.7 (Cq benzyl groups), 136.2, 135.8, 128.4, 127.9, 127.9, 127.9 (Ar), 126.1, 125.5 (=CH2), 104.5 (C-2′), 89.0 (C-1), 83.6, 81.9, 80.5, 79.7, 77.7, 77.2 and 69.2 (C-2,3,3′,4,4′,5,5′), 75.8, 74.9, 73.5, 73.3, 72.8, 71.9, 71.7 (C-1′ + 6 OCH2Ph), 64.2, 62.9 (C-6,6′), 18.4, 18.3 (CH3).
MALDI-TOF MS calcd C62H66O13K: [M]+ 1057.4140; found 1057.4150.
4.3. 6,6′-Di-O-allyl-1′,2,3,3′,4,4′-hexa-O-benzylsucrose 3
To a solution of 1′,2,3,3′,4,4′-hexa-O-benzylsucrose (1, 500 mg, 0.56 mmol) in DMF (15 mL) was added NaH (60% dispersion in mineral oil, 68 mg, 1.68 mmol) and a catalytic amount of imidazole (10 mg). The mixture was cooled to 0 °C in an ice bath and stirred with exclusion of moisture for 15 min, and then, allyl bromide (205 mg, 0.15 mL, 1.68 mmol) was added dropwise. The mixture was placed in the microwave cavity and subjected to MW irradiation of max 300 W at constant temperature 145 °C for 10 min. Excess of the hydride was destroyed by careful addition of water, and the mixture was partitioned between water and ether (50 mL each). The organic phase was washed with water, dried, concentrated, and the product 3 was isolated by column chromatography (hexane-EtOAc, 4 : 1) as a colorless oil (400 mg, 74%), [α]D20 + 30.2 (c 1.2, CHCl3); Lit.[Citation27] [α]D + 29.9 (c 1.0, CHCl3). Anal. calcd for C60H66O11: C, 74.82; H, 6.91. Found: C, 74.96; H 7.19. The compound was identical in all respects with the substance previously described in the literature.[Citation27]
4.4. Poly(1′,2,3,3′,4,4′-hexa-O-benzyl-6,6′-di-O-methacryloylsucrose)-co-polystyrene 4 and poly(6,6′-di-O-allyl-1′,2,3,3′,4,4′-hexa-O-benzylsucrose)-co-polystyrene, 5
To a water solution (100 mL), containing PVA 3% w/v and sodium persulfate, Na2S2O8 (1% with respect to the monomers mixture), was added toluene solution (10 mL) containing 2 (500 mg, 0.49 mmol) or 3 (500 mg, 0.52 mmol), and styrene (10 equiv, 0.56 mL, 4.9 mmol or 0.59 mL, 5.2 mmol). The mixture was sparged with Ar for 15 min, then heated at 70 °C for 24 h while stirred vigorously. After cooling, the resulted heterogeneous mixture was centrifuged, decanted, washed with distilled water and then cold methanol to remove any residual monomers. The residue was freeze-dried to yield 4 (403 mg, 39% from the monomers weight) or 5 (385 mg, 37% from the monomers weight) as insoluble amorphous powders. IR (4): νmax (KBr): 3027 (C–H, stretch, unsaturated), 2924 (C–H, stretch, saturated), 1733 (C = O), 1495, 1453 (C–C stretch), 1295 (C–H, assym.), 1072 (C–O–C; C–O–H, stretch), 1027 (C–H, bending), 735, 698 (Ar) cm−1. IR (5): νmax (KBr): 3024 (C–H, stretch, unsaturated), 2922 (C–H, stretch, saturated), 1600, 1492, 1451 (C–C stretch, Ar), 1071 (C–O–C; C–O–H, stretch), 1027 (C–H, bending), 757, 698 (Ar) cm−1.
Supporting Information Available
IR, NMR and mass spectra/ elemental analysis of all compounds; 1H-NMR of filtrates; IR and DSC thermograms of the polymers. This material is available free of charge via the Internet.
supplementry.pdf
Download PDF (2.3 MB)Acknowledgements
This work has been supported by Fundação para a Ciência e a Tecnologia through Grant No. PEst-C/EQB/LA0006/2013 and PTDC/SAU-BMA/122444/2010 (AFM equipment). The NMR spectrometers are part of The National NMR Facility, supported by Fundação para a Ciência e a Tecnologia (RECI/BBB-BQB/0230/2012).
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Brannon-Peppas L, Blanchette JO. Nanoparticle and targeted systems for cancer therapy. Adv. Drug Delivery Rev. 2004;56:1649–1659.10.1016/j.addr.2004.02.014
- Peça IN, Petrova KT, Cardoso MM, Barros MT. Preparation and characterization of polymeric nanoparticles composed of poly(DL-lactide-co-glycolide) and poly(DL-lactide-co-glycolide)-co-poly(ethylene glycol) 10% Triblock end-capped with a galactose moiety. React. Funct. Polym. 2012;72:729–735.
- Paul DR, Robeson LM. Polymer nanotechnology: nanocomposites. Polymer. 2008;49:3187–3204.10.1016/j.polymer.2008.04.017
- Buzea C, Blandino IP, Robbie K. Nanomaterials and nanoparticles: sources and toxicity. Biointerphases. 2007;2:MR17–MR172.
- Brouwer N, Weda M, van-Riet-Nales D, de-Kaste D. Nanopharmaceuticals: implications for the European pharmacopoeia. Pharmeuropa. 2010;22:5–7.
- Liu S, Maheshwari R, Kiick KL. Polymer-based therapeutics. Macromolecules. 2009;42:3–13.10.1021/ma801782q
- Liao SW, Yu T-B, Guan Z. De novo design of saccharide-peptide hydrogels as synthetic scaffolds for tailored cell responses. J. Am. Chem. Soc. 2009;131:17638–17646.10.1021/ja907097t
- Barros MT, Petrova KT. Ziegler–Natta catalysed polymerisation for the preparation of copolymers with pendant sucrose moieties. Eur. Polym. J. 2009;45:295–301.10.1016/j.eurpolymj.2008.10.013
- Petrova KT, Potewar TM, Ascenso OS, Barros MT. Amide-linked N-methacryloyl sucrose containing polymers. Carbohydr. Polym. 2014;110:38–46.10.1016/j.carbpol.2014.03.050
- Petrova KT, Correia-da-Silva P, Crucho CIC, Barros MT. Chemoselective synthesis of sucrose building blocks and their polymerization. Curr. Org. Chem. 2014;18:1788–1802.10.2174/1385272819666140527231535
- Singh RP, Pandey JK, Rutot D, Degée P, Dubois P. Biodegradation of poly(ε-caprolactone)/starch blends and composites in composting and culture environments: the effect of compatibilization on the inherent biodegradability of the host polymer. Carbohydr. Res. 2003;338:1759–1769.10.1016/S0008-6215(03)00236-2
- Galgali P, Varma AJ, Puntambekar US, Gokhale DV. Towards biodegradable polyolefins: strategy of anchoring minute quantities of monosaccharides and disaccharides onto functionalized polystyrene, and their effect on facilitating polymer biodegradation. Chem. Commun. 2002;2884–2885.10.1039/b209254a
- Tokiwa Y, Fan H, Hiraguri Y, Kurane R. Biodegradation of a sugar branched polymer consisting of sugar, fatty acid, and poly(vinyl alcohol). Macromolecules. 2000;33:1636–1639.10.1021/ma981451v
- Barros MT, Petrova KT, Singh RP. Synthesis and biodegradation studies of new copolymers based on sucrose derivatives and styrene. Eur. Polym. J. 2010;46:1151–1157.10.1016/j.eurpolymj.2010.02.002
- Barros MT, Petrova KT, Singh RP. Synthesis of hydrophilic and amphiphilic acryl sucrose monomers and their copolymerisation with styrene, methylmethacrylate and α- and β-pinenes. Int. J. Mol. Sci. 2010;11:1792–1807.10.3390/ijms11041792
- Queneau Y, Jarosz S, Lewandowski B, Fitremann J. Sucrose chemistry and applications of sucrochemicals. Adv. Carbohydr. Chem. Biochem. 2008;61:217–292.
- Patil DR, Dordick JS, Rethwisch D. Chemoenzymatic synthesis of novel sucrose-containing polymers. Macromolecules. 1991;24:3462–3463.10.1021/ma00011a068
- Poschalko A, Rohr T, Gruber H, et al. SUBPOL: a novel sucrose-based polymer support for solid-phase peptide synthesis and affinity chromatography applications. J. Am. Chem. Soc. 2003;125:13415–13426.10.1021/ja035874a
- Hou X, Yang J, Tang J, Chen X, Wang X, Yao K. Preparation and characterization of crosslinked polysucrose microspheres. React. Funct. Polym. 2006;66:1711–1717.10.1016/j.reactfunctpolym.2006.06.009
- Prasad K, Bahadur P, Meena R, Siddhanta AK. Facile solvent free synthesis of polymerised sucrose functionalised polyoxyethylene (23) lauryl ether by microwave irradiation. Green Chem. 2008;10:1288–1293.
- Ortiz RA, Martinez AYR, Valdez AEG, Duarte MLB. Preparation of a crosslinked sucrose polymer by thiol–ene photopolymerization using dithiothreitol as comonomer. Carbohydr. Polym. 2010;82:822–828.10.1016/j.carbpol.2010.05.054
- Jung S, Huh S, Cheon YP, Park S. Intracellular protein delivery by glucose-coated polymeric beads. Chem. Commun. 2009;2009:5003–5005.
- Barros MT, Petrova KT, Correia-da-Silva P, Esteves AP. Chapter 14, Efficient microwave-assisted synthesis of 1′,2,3,3′,4,4′,6-hepta-O-benzyl-sucrose and 1′,2,3,3′,4,4′-hexa-O-benzylsucrose. In: van-der-Marel G, Codee J, editors. Carbohydrate chemistry: proven synthetic methods. Vol. 2. Boca Raton (FL): Taylor & Francis Group: CRC Press; 2014. p. 103–123.
- Raposo CD, Petrova KT, Barros MT. Microwave-assisted protocols applied to the synthesis of 1′,2,3,3′,4,4′-hexa-O-benzylsucrose. Synth. Commun. 2014;44:3027–3036.10.1080/00397911.2014.926555
- Barros MT, Petrova KT, Correia-da-Silva P. Chapter 14, Sucrose chemistry: fast and efficient microwave-assisted protocols for the generation of sucrose-containing monomer libraries. In: Chandra U, editor. Microwave heating (ISBN 979-953-307-020-8). Vol. 1. Rijeka: InTech – Open Access Publisher; 2011. p. 309–332.
- Jarosz S, Listkowski A, Mach M. Coupling of the C6 and C6′ positions of sucrose by metathesis reaction. Pol. J. Chem. 2001;75:683–687.
- Lewandowski B, Listkowski A, Petrova KT, Jarosz S. Chapter 44, Functionalisation of terminal positions of sucrose – Part II: preparation of 1′,2,3,3′,4,4′-hexa-O-benzyl sucrose and 6,6′-bis-O-(2-hydroxyethyl)-1′,2,3,3′,4,4′-hexa-O-benzylsucrose. In: Kovac P, editor. Carbohydrate chemistry: proven synthetic methods. Vol. 1. Boca Raton (FL): Taylor & Francis Group, CRC Press; 2011. p. 407–425.
- Glavchev I, Petrova K, Devedjiev I. Determination of the rate of cure of epoxy resin/maleic anhydride/Lewis acids. Polym. Test. 2002;21:89–91.10.1016/S0142-9418(01)00053-8
- Glavchev I, Petrova K, Devedjiev I. Determination of the activity of phosphorus containing compounds on the crosslinking of epoxy resins by acid anhydrides. Polym. Test. 2002;21:243–245.10.1016/S0142-9418(01)00075-7
- Barros MT, Petrova K, Ramos AM. Regioselective copolymerization of acryl sucrose monomers. J. Org. Chem. 2004;69:7772–7775.10.1021/jo048957y
- Perrin DD, Armagedo WLF, Perrin DR. Purification of laboratory chemicals. 2nd ed. New York (NY): Pergamon Press Ltd; 1980.