
Abstract
In this work, we describe the design and synthesis of novel core-fluorinated Schiff base monomers and conjugated polymers based on them. The new fully aromatic highly fluorinated poly(azomethine ether)s (PAMEs) were prepared by polycondensation of core-fluorinated azomethine-containing compounds. The structure of the monomers and polymers were confirmed by FTIR, 1H, 13C, and 19F NMR spectroscopic analysis. The influence of synthesis condition on the properties of PAME compounds was investigated. Application of polarization microscopy with a temperature control thermal stage revealed thermotropic liquid crystalline (LC) behavior in the synthesized materials. Transition temperatures and a range of the existence of the LC phase were studied by a combination of the optical microscopy and DSC analysis. According to the TGA analysis, all the synthesized PAMEs show high thermal stability and thus offer a wide range of thermal processibility (up to 410–477 °C), which makes them prospective materials for many modern applications.
1. Introduction
Polyazomethines (PAMs) or polymeric Schiff bases are an interesting class of polymers and show various excellent properties such as good thermal and environmental stability, mechanical strength, nonlinear optical, and semiconducting electrical properties under doping. They often display liquid crystalline (LC) behavior at room temperature as well as ability to form chelates with metals.[Citation1–8] As a consequence, PAMs have a potential for numerous application in electronics, optoelectronics, photonics, and sensors.[Citation6–8]
PAMs can be synthesized in a traditional approach (also known as polyazomethine route) by a reaction of dialdehydes with diamines.[Citation4,9–16] Unfortunately, the produced polymers are characterized by limited solubility in the most of common organic solvents.[Citation3,4,6] The growing macromolecular chains of PAMs precipitate from the solution during the polycondensation reaction. As a result, high molecular weight polymers are very difficult to obtain under the traditional synthetic route. Owing to strong interchain interactions, the PAMs have limited solubility, which is considered as a major drawback associated with such class of polymers.[Citation17] Moreover, high melting temperatures of the PAMs make them intractable for processing by other conventional techniques.[Citation3,4,6,17]
One of possible ways for solving the aforementioned problem is in obtaining PAMs by an alternative route that consists in using of azomethine-containing compounds (like bis-diols, bis-phenols, etc.).[Citation4,18–21] The compounds synthesized in such way have an advantage of simplicity of their synthesis, which leads also to soluble polymer materials. Several types of PAMs like poly(azomethine ether)s (PAMEs), poly(azomethine urethane)s, and poly(azomethine imide)s have been recently synthesized using the alternative approach. Let us note that the polymer chains propagation reaction can occur via formation of ether, urethane, or imide bonds in this case.[Citation18–21]
It is generally recognized that the presence of alkyl chains and isomeric fragments in a polymer backbone imparts higher segmental mobility to the polymers and so simultaneously enhances their solubility and reduces their glass transition temperatures.[Citation22] Additionally, the incorporation of the aliphatic moieties and flexible ether linkages into the polymer chains of PAMs (in particular, in PAMEs) is sufficient to induce LC properties to such macromolecular compounds.[Citation3,6,23]
On the other hand, it is known that introduction of fluorine atoms into polymers’ structure enhances their thermal stability and chemical resistance, as well as allows reduction the polymers’ dielectric constant, the refractive index, optical losses at the same time improving their ability to form LC phases.[Citation24–30] It is interesting to note that such fluorinated liquid crystals have good stability, high voltage holding ratio, and specific resistance. Moreover, they are characterized with low threshold switching voltage (Vth) and small temperature coefficient of Vth, and are, therefore, widely used now in LC displays.[Citation24,26]
The aim of this study was in the synthesis of new advanced isomeric fluorinated PAMEs based on aromatic core-fluorinated dihydroxyl-substituted azomethine-containing monomers and bis(pentaflurophenyl)-derivatives having tetrafluorobenzene (TFB) or octafluorobiphenylene (OFB) central unit. The optimal synthesis conditions as well as physical (thermotropic LC behavior) and chemical (solubility, degradation stability) properties of the prepared fluorinated poly(azomethine ether)s were studied in a wide temperature range.
2. Experimental
2.1. Materials
Initial 4-[4-(4-aminophenoxy)-2,3,5,6-tetrafluorophenoxy]phenyl amine (1), 3-[4-(3-aminophenoxy)-2,3,5,6-tetrafluorophenoxy]phenyl amine (6), 3-{[4′-(3-aminophenoxy)-2,2′,3,3′,5,5′,6,6′-octafluoro-1,1′-biphenyl-4-yl]oxy}phenyl amine (7) were synthesized according to the previously reported procedure.[Citation31] Isomeric para-hydroxybenzaldehyde (2) (Sigma-Aldrich, 98%), meta-hydroxybenzaldehyde (3) (Sigma-Aldrich, 97%), p-toluenesulfonic acid (Merk, 98%), dibromopentane (11) (Merk, 97%), and dibromohexane (12) (Merk, 96%), pentafluorobenzaldehyde (8) (Sigma-Aldrich, 98%) were used without further purification. Other reagents and solvents were purified by well-established techniques.
2.2. Measurements
Fourier transform infrared (FTIR) spectra of synthesized compounds were recorded with a TENSOR 37 spectrometer in the absorption region of 600–4000 cm−1. 1H, 13C, and 19F NMR spectra were recorded on a Bruker Avance DRX 500 MHz and Bruker Avance 250 MHz spectrometers at room temperature with deuterated dimethyl sulfoxide (DMSO-d6) or chloroform (CDCl3) as standards. Chemical shifts are reported relative to DMSO-d6 (δ = 2.50 ppm) or CDCl3 (δ = 7.25 ppm) for 1H NMR. Fluorotrichloromethane or hexafluorobenzene were used as the internal standard for 19F NMR. UV–vis spectra were recorded with a Specord 210 (Analytikjena) spectrometer using dimethylformamide (DMF) solutions of compounds. Intrinsic viscosities [η] of polymer solutions were determined with an Ubbelohde viscometer in concentrated sulfuric acid or DMF at 30 °C. Gel permeation chromatography (GPC) of PAMEs was carried out on a Waters Breeze 1515 GPC system. Waters Styragel columns with tetrahydrofuran (THF) as an eluent were used in this study. Differential scanning calorimetry (DSC) was performed by a TA Instruments Q-2000 apparatus (United States) at a heating rate of 20 K/min. The thermo-oxidative destruction of polymers was studied via thermogravimetric analysis (TGA) on a TA Instruments Q-50 apparatus (United States) under air condition. A heating rate of 20 K/min within the temperature range from 25 to 700 °C was applied.
To inspect the existence and a temperature range of LC phases of the synthesized materials, we used an Olympus BX51 polarization microscope equipped with a temperature control system based on an HS1 precision hot stage (Instec, Inc.). For an accurate identification of the LC phase, the material has to be properly aligned. In this work, we used spin-coated and rubbed films of a commercial polyimide PI-2555 (Nissan Chemical Industries, Ltd.) to make in plane alignment of the nematic phase. A single orientation in the plane of the bounding substrate was determined by the rubbing direction of the polyimide sublayer. Glass slides were thoroughly washed using an Alconox detergent and D.I. water (for 10 min each in the ultrasonic bath at 60 °C) and then dried in air. For preparation of the aligning sublayer, PI-2555 polyimide and DMF solvent were mixed in the 1:1 ratio. The solution was cast onto the glass substrates by a spin-coater at 1500 rpm. The coatings were first dried at 100 °C for 2 min and then baked at 180 °C for 1 h. The polyimide-coated substrates were rubbed using an aluminum block covered with a cloth. The cells were assembled from pairs of the substrates with rubbing directions being anti-parallel to each other.
2.3. Monomer synthesis
4,4′-{(2,3,5,6-tetrafluoro-1,4-phenylene)bis[oxy-4,1-phenylenenitrilomethyl-ylidene]} diphenol (4)
The reactions were performed by setting diamine 1 (0.318 g, 0.873 mmol), para-hydroxybenzaldehyde 2 (0.213 g, 1.746 mmol), and para-toluenesulfonic acid (catalytic amount) into a round-bottom flask. Absolute ethanol (50 ml) was added into the flask, and the reaction mixture was stirred and heated under reflux for 22 h. Then, the mixture was cooled to room temperature. The precipitated monomer was filtered and washed with ethanol several times. The resulting product was purified by boiling in ethanol and dried under vacuum at 80 °C. Yield: 0.375 g (75%); m.p.: more than 250 °C. 1H NMR (DMSO-d6, 500 MHz, δ, ppm): 10.12 (s, 2H, –OH), 8.49 (s, 2H, –CH=N–), 7.78 (d, 4H, J = 8.3 Hz, Ph), 7.27 (d, 8H, J = 4.7 Hz, Ph), 6.89 (d, 4H, J = 8.3 Hz, Ph).13C NMR (DMSO-d6, 125.73 MHz, δ, ppm): 160.69, 159.92, 154.82, 147.96, 144.26, 130.73, 127.57, 122.48, 116.04, 115.72. 19F NMR (DMSO-d6, 188.14 MHz, δ, ppm): −154.28 (s, 4F, Ph). FTIR (v, cm−1): 3371–3293 (OH), 2925–2491 (CH), 1604 (–CH=N–), 1577, 1513–1492 (Ph), 1205–1187 (Ph–O–Ph), 1008–989 (C–F). UV–vis: λmax = 326 nm.
3,3′-{(2,3,5,6-tetrafluoro-1,4-phenylene)bis[oxy-4,1-phenylenenitrilomethyl-ylidene]- diphenol (5)
The reactions were performed by setting diamine 1 (0.318 g, 0.873 mmol) and meta-hydroxybenzaldehyde 3 (0.213 g, 1.746 mmol) into a round-bottom flask. Absolute ethanol (50 ml) was added into the flask, and the reaction mixture was stirred and heated under reflux for 5 h. Then, the mixture was cooled to room temperature. The precipitated monomer was filtered and washed with ethanol several times. The resulting product was purified by boiling in ethanol and dried under vacuum at 80 °C. Yield: 0.4 g (80%); m.p.: more than 250 °C. 1H NMR (DMSO-d6, 400 MHz, δ, ppm): 9.71 (s, 2H, –OH), 8.56 (s, 2H, –CH=N–), 7.37–7.27 (m, 14H, Ph), 6.94 (d, 2H, J = 6.6 Hz, Ph). 13C NMR (DMSO-d6, 125.73 MHz, δ, ppm): 160.56, 157.77, 155.23, 147.38, 137.46, 129.91, 122.72, 120.33, 118.80, 116.44, 114.77, 114.35. 19F NMR (DMSO-d6, 500 MHz, δ, ppm): −154.66 (s, 4F, Ph). FTIR (v, cm−1): 3401–3317 (OH), 2908–2495 (CH), 1625 (–CH=N–), 1591, 1510–1419 (Ph), 1201 (Ph–O–Ph), 1006–991 (C–F). UV–vis: λmax = 323 nm.
N-[(1E)-(pentafluorophenyl)methylene]-N-{3-[2,3,5,6-tetrafluoro-4-(3-{[(1E)-(pentafluorophenyl)methylene]amino}1phenoxy)phenoxy]phenyl}amine (9)
The reactions were performed by setting diamine 6 (1 g, 2.74 mmol) and pentafluorobenzaldehyde 8 (1.076 g, 5.49 mmol) into a round-bottom flask. Benzene (50 ml) was added into the flask, and the reaction mixture was stirred and heated under reflux for 6 h. Then, the mixture was cooled to room temperature. The precipitated monomer was filtered and washed with benzene several times. The resulting product was purified by precipitation from chloroform solution in hexane and finally dried under vacuum at 80 °C. Yield: 1.760 g (89%); m.p.: 198–200 °C. 1H NMR (CDCl3, 400 MHz, δ, ppm): 8.55 (s, 2H, –CH=N–), 7.41 (t, 2H, J = 7.9 Hz, J = 8.4 Hz, Ph), 6.96–6.93 (m, 4H, Ph), 6.85 (s, 2H, Ph). 13C NMR (CDCl3, 125.73 MHz, δ, ppm): 157.45, 152.66, 149.29, 144.78, 142.82, 138.54, 136.33, 130.26, 129.12, 115.08, 113.79, 108.47, 102.99. 19F NMR (CDCl3, 376.32 MHz, δ, ppm): −141.24–−141.41 (m, 4F, Ph), −148.72 (t, 2F, J = 24.3 Hz, Ph), −153.79 (s, 4F, Ph), −161.70 to −162.83 (m, 4F, Ph). FTIR (v, cm−1): 1648 (–CH=N–), 1594, 1525–1498 (Ph), 1122 (Ph–O–Ph), 1014–966 (C–F). UV–vis: λmax = 320 nm.
N-(3-{[2,2′,3,3′,5,5′,6,6′-octafluoro-4′-(3-{[(1E)-(pentafluorophenyl)methyl-ene]amino}henoxy)-1,1′-biphenyl-4-yl]oxy}phenyl)-N-[(1E)-(pentafluorophenyl)-methylene]amine (10)
The compound 10 was prepared in the same manner as 9, with diamine 7 used instead of diamine 6. Yield: 1.576 g (93%); m.p.: 199–203 °C. 1H NMR (CDCl3, 400 MHz, δ, ppm): 8.57 (s, 2H, –CH=N–), 7.43 (t, 2H, J = 8.42 Hz, J = 7.9 Hz, Ph), 7.01–6.97 (m, 4H, Ph), 6.90 (s, 2H, Ph). 13C NMR (CDCl3, 125.73 MHz, δ, ppm): 157.17, 152.68, 149.39, 147.91, 146.84, 145.39, 144.77, 143.33, 142.34, 140.33, 138.32, 136.43, 134.60, 130.32, 129.84, 115.31, 114.1, 110.7, 108.77, 102.4. 19F NMR (CDCl3, 188.14 MHz, δ, ppm): −137.82–−137.93 (m, 4F, Ph), −141.54 (d, 4F, J = 20.2 Hz, Ph), −149.51 (t, 2F, J = 20.1 Hz, Ph), −153.25 (d, 4F, J = 16.1 Hz, Ph), −161.7 to −162.0 (m, 4F, Ph). FTIR 1650 (–CH=N–), 1592, 1527–1490 (Ph), 1128 (Ph–O–Ph), 1018–970 (C–F). UV–vis: λmax = 318 nm.
2.4. Preparation of polymers
Synthesis of PAME-I
Bisphenol 4 (0.249 g, 0.435 mmol), potassium carbonate (0.072 g, 0.522 mmol) and 3 ml of dimethylacetamide (DMAc) were added into a three-necked round-bottom flask, which was fitted with a thermometer, magnetic stirrer, and argon gas supplier. The reaction mixture was heated up to 50 °C for 20 min and equivalent amount of 1.5-dibromopentane 11 (0.1 g, 0.435 mmol) was added into the flask. Then, reaction mixture was maintained at 100 °C for 48 h, cooled, and poured into 100 ml of water with stirring. A solid precipitate was isolated by filtration and thoroughly washed with water, hot ethanol, and finally dried under vacuum for 8 h at 80 °C. Yield: 0.234 g (84%). FTIR (v, cm−1): 2931–2869 (CH2), 1623 (–CH=N–), 1600, 1510–1490 (Ph), 1240–1209 (Ph–O–Ph), 1007–993 (C–F).
Synthesis of PAME-II
The PAME-II was prepared in the same manner as PAME-I, with compound 5 used instead of compound 4.
Yield: 0.217 g (78%); 1H NMR (CDCl3, 500 MHz, δ, ppm): 8.42 (s, 2H, –CH=N–), 7.51 (s, 2H, Ph), 7.38 (s, 4H, Ph), 7.22 (s, 4H, Ph), 7.05 (s, 6H, Ph), 4.09 (s, 4H), 1.91 (s, 4H), 1.66 (d, 2H). FTIR (v, cm−1): 3097–2869 (CH2), 1625 (–CH=N–), 1606, 1496–1473 (Ph), 1243–1207 (Ph–O–Ph), 998–978 (C–F).
Synthesis of PAME-III
The PAME-III was prepared in the same manner as PAME-I, with compound 12 used instead of compound 11.
Yield: 0.212 g (79%); FTIR (v, cm−1): 2923–2854 (CH2), 1630 (–CH=N–), 1592, 1510 (Ph), 1251–1236 (Ph–O–Ph), 989 (C–F).
Synthesis of PAME-IV
The PAME-IV was prepared in the same manner as PAME-I, with compounds 5 and 12 used instead of compounds 4 and 11.
Yield: 0.202 g (75%); 1H NMR (CDCl3, 500 MHz, δ, ppm): 8.42 (s, 2H, –CH=N–), 7.51 (s, 2H, Ph), 7.37 (d, 4H, J = 7.7 Hz, Ph), 7.23 (d, 4H, J = 8.2 Hz, Ph), 7.04 (d, 6H, J = 7.7 Hz, Ph), 4.07 (s, 4H), 1.87 (s, 4H), 1.59 (s, 4H). FTIR (v, cm−1): 3064–2869 (CH2), 1627 (–CH=N–), 1596, 1506–1477 (Ph), 1249–1218 (Ph–O–Ph), 1003–978 (C–F).
Synthesis of PAME-V
Compound 4 (0.248 g, 0.433 mmol), potassium carbonate (0.072 g, 0.520 mmol), and DMF (3 ml) were added into a three-necked round-bottom flask, which was fitted with a thermometer, magnetic stirrer, and argon gas supplier. Reaction mixture was heated up to 50 °C for 20 min and equivalent amount of compound 9 (0.312 g, 0.433 mmol) was added into the flask. Then, reaction mixture was maintained at 80 °C for 20 min, and then poured into 100 ml of water with stirring. Produced solid precipitate was isolated by filtration and thoroughly washed with water, hot ethanol, and finally dried under vacuum for 8 h at 80 °C. Yield: 0.374 g (69%); FTIR (v, cm−1): 2964–2846 (CH2), 1639 (–CH=N–), 1594, 1508–1486 (Ph), 1272–1216 (Ph–O–Ph), 1004–975 (C–F).
Synthesis of PAME-VI
The polymer PAME-VI was prepared in the same manner as PAME-V, with compound 5 used instead of compound 4. The reaction mixture was stirred under a nitrogen stream at 80 °C for 15 min. Yield: 0.331 g (61%); FTIR (v, cm−1): 2927–2850 (CH2), 1633 (–CH=N–), 1592–1581, 1510–1475 (Ph), 1270–1205 (Ph–O–Ph), 987 (C–F).
Synthesis of PAME-VII
The polymer PAME-VII was prepared in the same manner as PAME-V, with compound 10 used instead of compound 9. Yield: 0.394 g (65%); FTIR (v, cm−1): 2933–2869 (CH2), 1625 (–CH=N–), 1604, 1494–1475 (Ph), 1243–1186 (Ph–O–Ph), 997–977 (C–F).
Synthesis of PAME-VIII
The polymer PAME-VIII was prepared in the same manner as PAME-VI, with compound 10 used instead of compound 9. Yield: 0.401 g (66%); FTIR (v, cm−1): 2968–2823 (CH2), 1623 (–CH=N–), 1598, 1510–1490 (Ph), 1236–1186 (Ph–O–Ph), 1006–991 (C–F).
3. Results and discussion
In order to prepare fluorinated PAMEs, core-fluorinated azomethine-containing monomers were first prepared. Dihydroxyl-substituted azomethine-containing monomers 4 and 5 with TFB unit were synthesized by a condensation reaction between diamine 1 and isomeric hydroxybenzaldehyde 2 or 3 (Scheme ).
Similarly, bis(pentafluorophenyl)azomethine-containing monomers with TFB (monomer 9) and OFB (monomer 10) central unit were obtained by interaction of core-fluorinated diamines 6 and 7 with pentafluorobenzaldehyde 8, respectively, (Scheme ).
The structure of the synthesized azomethine-containing monomers 4, 5, 9, and 10 was confirmed by FTIR, 1H, 13C, 19F NMR, and UV–vis spectrometry techniques (see the Supporting Information).
We used the alternative approach (specifically polyether route) in order to synthesize azomethine-containing polymers (such as PAMEs) based on the obtained monomers. In this case, azomethine groups were already incorporated into the starting monomers.
The novel azomethine-containing PAME-I–PAME-IV polymers having aliphatic moiety were prepared by polycondensation reactions of monomers 4 and 5 with dibromoalkanes 11 and 12 (Scheme ).
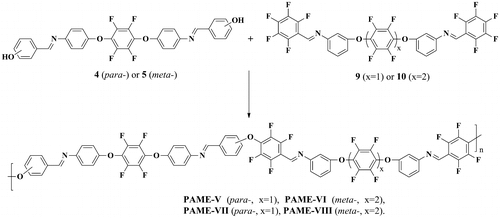
The fully aromatic PAME-V–PAME-VIII were prepared by polycondensation of the compound 4 with bis(pentafluorophenyl)azomethine-containing monomer 9 or 10 as well as of the compounds 5 with monomer 9 or 10 (Scheme ).
The conditions for obtaining PAMEs were found by changing the temperature and time of the reaction and solvent/monomer concentrations in the reaction medium. Polymers PAME-I–PAME-VIII were synthesized with the maximum yields and the highest [η] values by using anhydrous potassium carbonate as a base and monomer concentration in the reaction medium of 10%. Other synthesis conditions as well as yields and [η] values for PAMEs are listed in Table .
Table 1. The synthesis conditions, molecular weight properties, and yields for PAMEs.
It is worth noting that the application of bis(pentaflurophenyl)azomethine-containing derivatives 9 and 10 allowed synthesis of PAMEs under relatively mild conditions as can be seen from Table . This result indicates that the fluorine atom in a para-position in pentafluorophenyl unit is very reactive with the activated dihydroxyl-substituted monomers. However, the fluorine atoms at the ortho-position of the pentafluorophenyl unit can also react under appropriate conditions with dihydroxyl-substituted compounds. Thus, an increase in the reaction temperature or an extended reaction time yields cross-linked products.
The resulting polymers PAME-I, PAME-III, PAME-V, and PAME-VII are soluble only in the concentrated sulfuric acid, while PAME-II and PAME-IV are soluble in most common organic solvents and insoluble in methanol, ethanol, toluene, benzene, and n-hexane. Exception are polymers PAME-VI and PAME-VIII, which are soluble only in the solvents with a high boiling point such as DMSO, NMP, DMAc under heating and are precipitating from of the solutions during consequent cooling. The improved solubility of the polymers, PAME-II, PAME-IV, in polar organic solvents may be attributed to the simultaneous incorporation of isomeric fragments and flexible aliphatic segments into the polymer chains.[Citation22,32]
The solubility of PAME-II and PAME-IV polymers in THF enabled investigation of their molecular mass distribution by GPC using polystyrene standards. According to the GPC measurements, the PAME-IV polymer based on 1.6-dibromohexane has higher molecular weight than the PAME-II compound based on the 1.5-dibromopentane unit. The Mn values of PAME-II and PAME-IV are 3400 and 7600 g/mol, respectively. The Mw/Mn values of these polymers were similar: 2.9 for PAME-II and 3.2 for PAME-IV (Table ).
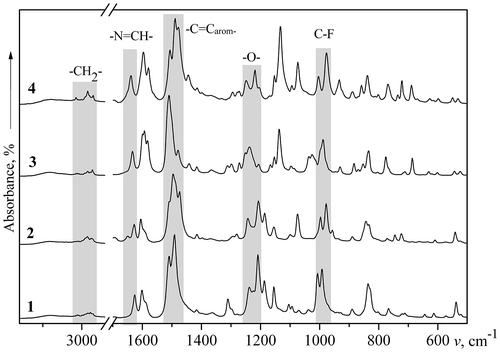
FTIR spectra of the obtained polymers show no sign of an intense broad band in the range of 3600–3100 cm−1 corresponding to OH groups and characteristic for the initial monomers 4 and 5. This indicates that these groups have been consumed during the polycondensation reaction. Additionally, FTIR spectra of all PAMEs show peaks in the regions 976–1007 cm−1, 1200–1254 cm−1, 1475–1510 cm−1, 1623–1630 cm−1, and 2800–3100 cm−1, which correspond to C–F, –O–, –C=Carom−, –N=CH–, and –CH2– groups, respectively. FTIR spectra of PAME-I–PAME-IV are presented in Figure . FTIR spectra of the fully aromatic PAMEs are given in the Supporting Information.
Figure 1. FTIR spectra of polymers PAME-I (curve 1), PAME-II (curve 2), PAME-III (curve 3), and PAME-IV (curve 4).
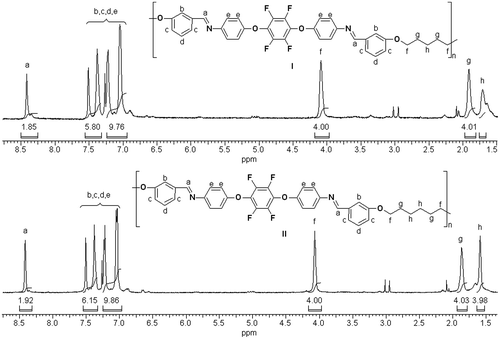
The 1H NMR spectra of the soluble polymers (PAME-II and PAME-IV) in CHCl3 are illustrated in Figure . All the observed peaks could be readily assigned to the protons in the corresponding repeat units. 1H NMR spectra of the synthesized polymers do not show any signals corresponding to the terminal –OH groups of the initial monomers 4 and 5. Also, the 1H NMR spectra PAME-II and PAME-IV exhibit a signal at 8.42 ppm (a) indicating the presence of protons of the –HC=N– group, whereas the resonance in the range 7.04–7.51 ppm indicates the presence of aromatic protons (b, c, d, and e). In addition to these signals, which were common in all the spectra, the polymers having aliphatic moiety show singlet peaks (f, g, h) in the range 1.66–4.09 ppm (aliphatic –CH2).
Figure 2. 1H NMR spectra of PAME-II (I) and PAME-IV (II).
Polymers PAME-I–PAME-IV were also synthesized using the traditional approach (polyazomethine route). Thus, PAMEs (marked as PAME-Ia–PAME-IVa) with the chemical structure identical to PAME-I–PAME-IV polymers were obtained by the interaction of the initial fluorine-containing diamine 1 with appropriate dibenzaldehydes having aliphatic moieties. Experimental details and characterization data (FTIR and 1H NMR spectra, inherent viscosities, and GPC measurements) for the synthesized PAME-Ia–PAME-IVa are presented in the Table and Supporting Information. As expected, the polymers prepared through the traditional route showed lower [η] and Mn values compared to PAME-I–PAME-IV prepared by the alternative route. Due to this fact, the polymers PAME-Ia–PAME-IVa were not studied further in this work.
Phase transition temperatures for all the polymers were investigated by DSC analysis. The DSC thermograms of PAME-I–PAME-IV are displayed in Figure ; also, the characteristic temperatures of PAME-I–PAME-IV and fully aromatic PAMEs (PAME-V–PAME-VIII) are listed in Table . As can be observed, polymers with similar substitution show similar thermal behavior (i.e., PAME-I and PAME-II vs. PAME-III and PAME-IV).
Figure 3. DSC thermograms of polymers PAME-I (curve 1), PAME-III (curve 2), PAME-II (curve 3), and PAME-IV (curve 4).
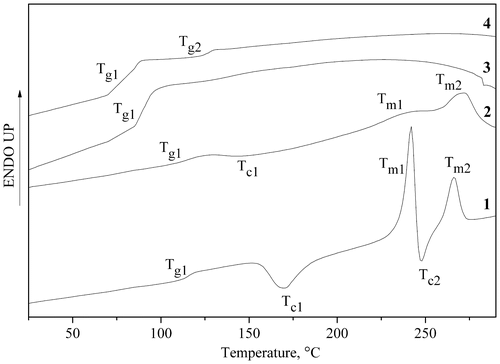
Table 2. Thermal behavior of the PAMEs.
Interestingly, the DSC thermograms of aliphatic-containing PAMEs with a para-azomethine substituent in the benzene ring (PAME-I and PAME-III) show three endothermic peaks in the second heating run (Figure and Table ). The first peak at lower temperature corresponds to the Tg value of the polymers (Table ). The other two peaks can be attributed to a presence of at least one LC phase.[Citation33–36] We believe that the second and the third endothermic peak of PAMEs are related to the melting (crystal-liquid Tm1) and isotropization transitions (Tm2), respectively, (see Figure and Table ).[Citation34] The DCS thermograms of PAME-I and PAME-III polymers have an additional exothermic peak above the glass transition temperature (presumably crystallization transition Tc1) indicating formation of ordered structures due to a cold crystallization process above Tg (Figure ). In addition, the melting transition Tm1 at 242 °C is followed by the second crystallization peak (Tc2) in these polymers. Due to insolubility of aliphatic-containing PAMEs with para-azomethine substituent in the benzene ring in common organic solvents, the corresponding LC properties were not studied in this work.
DSC studies revealed the amorphous nature of the aliphatic-containing PAME-II and PAME-IV with a meta-azomethine substituent in the benzene ring (Figure ). It is known that meta-substitution may reduce coplanarity of aromatic main-chain polymers, thus decreasing their ability to form crystalline domains.[Citation37,38] Our DSC studies revealed only one transition (Tg) in the PAME-II polymer. On contrary, the heat capacity curve recorded for the polymer PAME-IV (based on 1.6-dibromohexane) clearly displays two glass transitions (Figure and Table ).
The difference may be attributed to the existence of two amorphous phases in the latter compound. The first transition (Tg1) corresponds to a mobile phase constituted mainly with aliphatic units, while the second one (Tg2) should be attributed to relatively rigid amorphous phase, which includes aromatic fragments. Let us note that the PAME-IV based on 1.6-dibromohexane has higher Mn and correspondingly higher Tg value in comparison with the PAME-II polymer based on 1.5-dibromopentane.
It should be mentioned that no endothermic melting peaks common for LC polymers could be observed in DSC thermograms of PAME-II and PAME-IV on the heating ramp. However, it is well known that molecular reorientation in LC polymers depends on a previous heat treatment (thermal history) and, therefore, in some cases, the endothermic melting peaks can be found only after a proper annealing of a polymer.[Citation39–41] Polarization microscopy techniques allowed investigation of LC phases after long-term relaxation of the materials and achieving of the equilibrium in them. Figure displays representative polarization microscopy images recorded for a higher molecular weight polymer (PAME-IV) showing a well-pronounced thermotropic LC behavior typical for all polymers under study. The sample was annealed at 130 °C (in the isotropic melt state) for 2 h before measurements to erase its thermal history and was thermally stabilized at each temperature for 2 h before polarization microscopy characterization. The LC phase appeared in this polymer when it was heated above its glass transition temperature. In that state, the illumination light passed through the sample when it was viewed between the crossed polarizers revealing a uniform texture as a signature of an ordered structure. The observed ordering is similar to that typically exhibited by nonpolymeric liquid crystals. Further increase of temperature resulted in disappearance of the birefringence within the sample, which demonstrated the transition into an isotropic state.[Citation42] The established phase transition temperature, TLC-I, for the PAME-IV sample was about 88 °C. Further insight into the LC behavior could be given by the crossed polaroid texture observation (Figure (b) and (c)). It should be noted, however, that a traditional texture commonly observed for low molecular weight compounds can be more complicated in case of their polymeric counterparts. The low molecular mass LC materials possess much lower melt viscosities that enable relatively fast appearance of identifiable LC structures. Opposite to that, in highly viscose polymeric materials, the appearance of a distinct texture can take much longer time (from several minutes to several hours). This can lead to considerable difficulties and uncertainties in LC phases identification.[Citation43]
Figure 4. Polarization microscopy textures observed in PAME-IV polymer sandwiched between two glass slides with rubbed polyimide aligning coatings at 80 °C (a–b) and 95 °C (c); (a) and (b) images differ in the orientation of the average nematic director, n, with respect to polarizer (P) and analyzer (A) directions; (c) displays a characteristic negative tactoid texture observed in a biphasic LC-isotropic region.

Figure (c) gives an evidence of existence an additional anisotropic LC phase observed in the PAME-IV sample in the melt (T = 95 °C). The observed microscopy texture shows formation of negative tactoids, which are inclusions of the isotropic phase within the LC surrounding.[Citation44] These inclusions are of an elongated cusped shape, which is markedly different from the similar inclusions of spherical shape observed for a completely isotropic medium. The shape of the negative tactoids is determined by a balance of the anisotropic surface tension and LC elasticity of the region outside the inclusions.[Citation45]
According to DSC investigations, the thermal behavior of fully aromatic PAME-V–PAME-VIII is similar to PAMEs with meta-azomethine phenyloxy units. A single Tg value is found for PAME-V, whereas two glass transitions can be seen in DSC thermograms of PAME-VI, PAME-VII, and PAME-VIII (Table ). Evidently, two Tg values as well as wide range of ∆Tg (PAME-V–PAME-VIII polymers) might result from a large polydispersity and, probably, higher ability of these polymers to create two-dimensional assemblies with an aromatic ππ stacking interactions.[Citation46]
Generally, among the synthesized polymers, the fully aromatic PAME-V–PAME-VIII show the highest Tg values due to the presence of highly rigid aromatic units in the polymer backbone, confirmed with the smallest ΔCp values for these polymer (Table ). It is well known that appearance of LC phase in a polymer is connected to its chemical structure. Typical main-chain LC polymers have a rigid (several benzene rings) aromatic fragments (so-called mesogenic groups) as well as flexible (aliphatic) spacers between them. The length of the spacer controls the degree of coupling between the mesogenic groups, thus stabilizing the LC phase and prohibiting normal crystallization behavior. The latter may appear under conditions of effective decoupling of the mesogenic groups from the rest of the polymer chains (when the spacer is too long). In the opposite extreme case (small spacer or no spacer), the polymer chains may be too rigid to achieve a proper alignment within a reasonable time frame and will display LC ordering only in a solution state and under uniaxial drawing conditions. This general consideration is in tune with the structure of polymer materials under study. Fully aromatic fragments with TFB or OFB central units act as mesogenic groups, however, depending on the isomeric substitution, their role in the formation of ordered LC structures may vary. The difference in thermal behavior between fully aromatic and aromatic–aliphatic polymers is well pronounced. The latter have better solubility and lower Tg, while the former are insoluble and highly rigid (Tg > 138 °C). The difference between polymers having different spacer length is minor but also well pronounced. Obviously, the two aliphatic chain lengths are too similar to expect observation of the decoupling influence of the spacer length. However, the observed well-pronounced trend in the reduction of the Tg can be clear observed when comparing polymers PAME-I and PAME-III or PAME-II and PAME-IV (Table ).
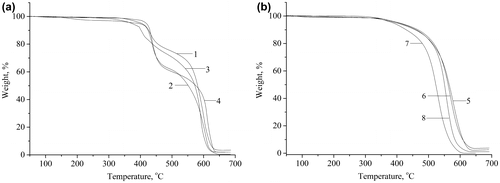
TGA curves of the aliphatic-containing PAMEs (PAME-I–PAME-IV) and fully aromatic PAME-V–PAME-VIII are shown in Figure . The temperature of a 5% weight loss (T5) and the temperature of a 10% weight loss (T10) are given in Table for the polymers under study. The T10 value is one of the main criteria to determine the relative thermal stability of a polymer. Examination of the data demonstrates that the T10 values of PAMEs are in the range of 410–477 °C (i.e., have good thermal stability). The highest thermal stability is observed for the PAME-VIII polymer, which contains the OFB fragments. It is noteworthy also that all fully aromatic PAMEs (PAME-V–PAME-VIII) show a one-step thermal decomposition Figure . On the contrary, in accordance with the TGA analysis all aliphatic-containing PAMEs (PAME-I–PAME-IV) are characterized with a two-step weight loss process upon heating (Figure ). The first step in the thermograms can be obviously ascribed to thermal decomposition of the aliphatic units. The second (higher temperature) degradation stage is most probably associated with decomposition of the ether and azomethine bonds.
Figure 5. TGA curves of polymers PAME-I (curve 1), PAME-II (curve 2), PAME-III (curve 3), and PAME-IV (curve 4), PAME-V (curve 5), PAME-VI (curve 6), PAME-VII (curve 7), and PAME-VIII (curve 8).
Scheme 1. Synthesis of core-fluorinated dihydroxyl-substituted azomethine-containing monomers 4 and 5.
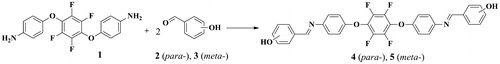
Scheme 2. Synthesis of bis(pentaflurophenyl)azomethine-containing derivatives 9 and 10.
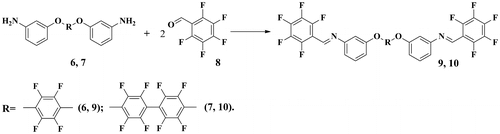
Scheme 3. Synthesis of core-fluorinated PAME-I–PAME-IV.
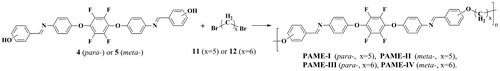
Scheme 4. Synthesis of fluorinated PAME-V–PAME-VIII.
One should note a considerable difference between the glass transition and the initial decomposition temperature in the polymers. This feature allows a wide processing window for the PAMEs under study offering an additional advantage for their commercial application.
4. Conclusion
In a summary, we have synthesized new core-fluorinated isomeric dihydroxyl-substituted azomethine-containing monomers with a tetrafluorobenzene central unit and bis(pentafluorophenyl)azomethine-containing monomers with perfluorinated mono- and biphenylene central nucleus. Novel polymers based on these new core-fluorinated compounds were also prepared. Our study of physical and chemical properties of the synthesized polymers shows that their solubility is mainly dependent on the isomeric nature of the PAMEs backbone. The thermal behavior of the synthesized polymers and especially the appearance of an LC phase in them are mainly determined by the core-fluorinated fragments of the polymers. Our studies demonstrate high thermal stability of the new PAMEs. It is reasonable to suggest that the products of PAMEs series with a meta-azomethine substituent in the benzene ring have good prospects for usage as main-chain thermotropic LC polymers. Due to additional possibility of doping PAME compounds to enable their electrical, optical, and sensor properties, the synthesized polymers may be interesting for many practical applications. These special properties of the core-fluorinated aromatic polymers require individual consideration will be a subject of our future work.
Supplemental data
The supplemental data for this article is available online at http://dx.doi.org/10.1080/15685551.2015.1092007.
Supplementary_material.pdf
Download PDF (795.5 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Kobzar Y, Tkachenko I, Shekera O, et al. The fluorine-containing polyazomethines: synthesis and properties. Polym. J. 2014;36:331–340.
- Pron A, Rannou P. Processible conjugated polymers: from organic semiconductors to organic metals and superconductors. Prog. Polym. Sci. 2002;27:135–190.
- Grigoras M, Catanescu CO. Imine oligomers and polymers. J. Macromol. Sci. Part C Polym. Rev. 2004;44:131–173.
- Iwan A, Sek D. Processible polyazomethines and polyketanils: from aerospace to light-emitting diodes and other advanced applications. Prog. Polym. Sci. 2008;33:289–345.
- Kumar S, Dhar DN, Saxena P. Applications of metal complexes of Schiff bases – a review. J. Sci. Ind. Res. 2009;68:181–187.
- Hussein MA, Abdel-Rahman MA, Asiri AM, et al. Review on: liquid crystalline polyazomethines polymers. Basics, syntheses and characterization. Des. Monomers Polym. 2012;15:431–463.
- Zhang W, Wang C, Liu G, et al. Structural effect on the resistive switching behavior of triphenylamine-based poly(azomethine)s. Chem. Commun. 2014;50:11496–11499.
- Ravikumar L, Kalaivani S, Vidhyadevi T, et al. Synthesis, characterization and metal ion adsorption studies on novel aromatic poly(azomethine amide)s containing thiourea groups. Open J. Polym. Chem. 2014;04:1–11.
- Saegusa Y, Kuriki M, Nakamura S. Preparation and characterization of fluorine-containing aromatic condensation polymers, 4. Preparation and characterization of fluorine-containing aromatic polyazomethines and copolyazomethines from perfluoroisopropylidene group-containing aromatic diamines and/or isopropylidene group-containing aromatic diamines and phthaladehydes. Macromol. Chem. Phys. 1994;195:1877–1889.
- Kumar Gutch P, Banerjee S, Gupta D, et al. Poly‐Schiff bases. V. Synthesis and characterization of novel soluble fluorine‐containing polyether azomethines. J. Polym. Sci., Part A: Polym. Chem. 2001;39:383–388.
- Grimm B, Krüger R-P, Schrader S, et al. Molecular structure investigations on fluorine containing polyazomethines by means of the MALDI–TOF–MS technique. J. Fluorine Chem. 2002;113:85–91.
- Choi E-J, Ahn J-C, Chien L-C, et al. Main chain polymers containing banana-shaped mesogens: synthesis and mesomorphic properties. Macromolecules. 2004;37:71–78.
- K-i Fukukawa, Shibasaki Y, Ueda M. Photosensitive poly (benzoxazole) via poly (o-hydroxy azomethine) II. Environmentally benign process in ethyl lactate. Polym. J. 2004;36:489–494.
- Krebs FC, Jørgensen M. The effect of fluorination in semiconducting polymers of the polyphenyleneimine type. Synth. Met. 2004;142:181–185.
- Ishii J, Tanaka Y, Hasegawa M. Film properties of polyazomethines (2). Poly(imide-azomethine)s derived from ester-containing dialdehydes, tetracarboxylic dianhydrides, and a fluorinated diamine. High Perform. Polym. 2010;22:145–158.
- Cerrada P, Oriol L, Pinol M, et al. Alternative synthetic strategies for Schiff base derived liquid crystal polymers: a comparative study. J. Polym. Sci., Part A: Polym. Chem. 1996;34:2603–2611.
- Stefanache A, Balan M, Harabagiu V, et al. Electro-optical properties of aromatic oligoazomethine/permethylated α-cyclodextrin main-chain polyrotaxanes. Chem. Phys. Lett. 2014;599:104–109.
- Tamareselvy K, Venkatarao K, Kothandaraman H. Synthesis and characterization of new halogen-containing poly (azomethine-urethane) s. Die Makromolekulare Chemie. 1990;191:1231–1242.
- Gauderon R, Plummer CJ, Hilborn JG, et al. Poly(arylene ether azomethine)s: synthesis by aldimine-activated nucleophilic aromatic substitution, characterization, and thin films morphological study. Macromolecules. 1998;31:501–507.
- Butt MS, Akhter Z, Zafar-uz-Zaman M, et al. Synthesis and characterization of some Schiff-base-containing polyimides. Colloid Polym. Sci. 2008;286:1455–1461.
- Kausar A, Zulfiqar S, Ahmad Z, et al. Novel processable and heat resistant poly(phenylthiourea azomethine imide)s: synthesis and characterization. Polym. Degrad. Stab. 2010;95:1826–1833.
- Gul A, Akhter Z, Siddiq M, et al. Synthesis and physicochemical characterization of poly (azomethine) esters containing aliphatic/aromatic moieties: electrical studies complemented by DFT calculation. J. Appl. Polym. Sci. 2014.
- Abid KK, Al-barody SM. Synthesis, characterisation and liquid crystalline behaviour of some lanthanides complexes containing two azobenzene Schiff base. Liq. Cryst. 2014;1–12.
- Ishikawa H, Toda A, Okada H, et al. Relationship between order parameter and physical constants in fluorinated liquid crystals. Liq. Cryst. 1997;22:743–747.
- Ma H, Jen AK-Y, Dalton LR. Polymer-based optical waveguides: materials, processing, and devices. Adv. Mater. 2002;14:1339–1365.
- Krishnan S, Kwark YJ, Ober CK. Fluorinated polymers: liquid crystalline properties and applications in lithography. Chem. Record. 2004;4:315–330.
- Smith JA, DiStasio RA, Hannah NA, et al. SF 5 -terminated fluorinated Schiff base liquid crystals. J. Phys. Chem. B. 2004;108:19940–19948.
- Shevchenko V, Tkachenko I, Shekera O. Nucleus-fluorinated aromatic polyethers. Polym. Sci. Ser. B. 2010;52:408–430.
- Tkachenko I, Shekera O, Bliznyuk V, et al. Fluorinated allyl-, acetyl-, and bromo-containing hydroxyl-substituted phenyl ethers with a hexafluorobenzene or decafluorobiphenyl central unit. J. Fluorine Chem. 2013;149:36–41.
- Tkachenko I, Shekera O, Shevchenko V. Allyl-containing polyaryl ethers with perfluorinated mono-and biphenylene fragments. Polym. Sci. Ser. B. 2013;55:336–343.
- Shevchenko V, Sidorenko A, Bliznyuk V, et al. Synthesis and properties of hydroxylated core-fluorinated diamines and polyurethanes based on them with azobenzene nonlinear optical chromophores in the backbone. Polymer. 2013;54:6516–6525.
- Kaya İ, Avcı A, Kolcu F, et al. Synthesis, characterization, optical, and electrochemical properties of thermal stable novel poly(azomethine-ether)s. Des. Monomers Polym. 2014;17:481–490.
- Cozan V, Gaspar M, Butuc E, et al. Azomethine sulfone macromers with thermotropic liquid crystalline behavior. Eur. Polym. J. 2001;37:1–8.
- Kannan P, Raja S, Sakthivel P. Synthesis and characterization of thermotropic liquid crystalline poly(azomethine ether)s. Polymer. 2004;45:7895–7902.
- Choi E, Ahn H-K, Lee JK, et al. Liquid crystalline twin epoxy monomers based on azomethine mesogen: synthesis and curing with aromatic diamines. Polymer. 2000;41:7617–7625.
- Iwan A, Palewicz M, Sikora A, et al. Aliphatic–aromatic poly(azomethine)s with ester groups as thermotropic materials for opto(electronic) applications. Synth. Met. 2010;160:1856–1867.
- Teoh MM, Chung T-S, Schiraldi DA, et al. Thin-film polymerization and ‘RIS’ Metropolis Monte Carlo simulation of fluorinated aromatic copoly(ester–amide)s. Polymer. 2005;46:3914–3926.
- Yang C-P, Su Y-Y, Hsu M-Y. Syntheses and properties of fluorinated polyamides and poly(amide imide)s based on 9,9-bis[4-(4-amino-2-trifluromethylphenoxy)phenyl]fluroene, aromatic dicarboxylic acids, and various monotrimellitimides and bistrimellitimides. Colloid Polym. Sci. 2006;284:990–1000.
- Sauer BB, Kampert WG, McLean RS. Thermal and morphological properties of main chain liquid crystalline polymers. Polymer. 2003;44:2721–2738.
- Grell M, Bradley DD, Inbasekaran M, et al. A glass-forming conjugated main-chain liquid crystal polymer for polarized electroluminescence applications. Adv. Mater. 1997;9:798–802.
- Wendorff J, Finkelmann H, Ringsdorf H. Structure and morphology of liquid‐crystalline polymers. J. Polym. Sci. Polym. Symp. Wiley Online Library; 1978. p. 245–261.
- Vroege GJ, Lekkerkerker H. Phase transitions in lyotropic colloidal and polymer liquid crystals. Rep. Prog. Phys. 1992;55:1241.
- Lenz RW. Characterization of thermotropic liquid crystalline polymers. Pure Appl. Chem. 1985;57:977–984.
- Nastishin YA, Liu H, Schneider T, et al. Optical characterization of the nematic lyotropic chromonic liquid crystals: light absorption, birefringence, and scalar order parameter. Phys. Rev. E. 2005;72:041711.
- Kim Y-K, Shiyanovskii SV, Lavrentovich OD. Morphogenesis of defects and tactoids during isotropic–nematic phase transition in self-assembled lyotropic chromonic liquid crystals. J. Phys.: Condens. Matter. 2013;25:404202.
- Hwang J, Li P, Carroll WR, et al. Additivity of substituent effects in aromatic stacking interactions. J. Am. Chem. Soc. 2014;136:14060–14067.