
Abstract
Di-block copolymers of poly(ethylene oxide) (PEO) and glycopolymers at three different pendant spacer lengths of glucose moiety were synthesized by deacetylation of pendant moieties of 2,3,4,6-tetra-O-acetyl-d-glucopyranosides which were prepared by reversible addition-fragmentation chain-transfer (RAFT) process. The water soluble deacetylated copolymers, PEO-b-poly(acryl-d-glucopyranoside) (PEO-b-PAG, 2a), PEO-b-poly[4′-(acryloxy)butyl-d-glucopyranoside] (PEO-b-PABG, 2b), and PEO-b-poly[6′-(acryloxy)hexyl-d-glucopyranoside] (PEO-b-PAHG, 2c) were characterized in comparison with acetylated copolymers by spectroscopic and gel permeation chromatography methods. In order to assess the biocompatibility of these three di-block copolymers, the adhesion (%), viability (%), and proliferation assays were carried out in MC3T3 cells in-vitro at different concentrations from 10 nM to 1000 μM. Qualitative analysis of cell cytoskeletal organization was obtained by immunostaining with talin and integrin α5. It was clearly indicative of well spreading of cells in the presence of all glycopolymers at lower concentrations but with an increase in the concentration (10 and 100 μM) showed significant change in the cell surface morphologies, despite having good cell adhesion and viability as compared to the control conditions. The use of di-block glycolpolymers at ≤ 100 μM of the concentration showed very good osteoblast cells adhesion and viability response; however at higher concentrations (1000 μM) of glycopolymers > 80% of cells lost their viability.
Introduction
The development of biologically responsive materials and coatings similar to the natural systems with desired performance of cells at in-vitro and in-vivo conditions is required to increase the biocompatibility by which to meet the need of continuously rising demand of implants.[Citation1,2] In the past, a range of materials including metals, ceramics, composites and polymers were developed [Citation3–17] and subsequently various modification methods were employed to tailor their structural and functional properties.[Citation18–29] Recently, the use of value-added glycoconjugates or glycopolymers – characteristic synthetic macromolecules with pendant carbohydrate moieties – has found applications as materials for cell/tissue engineering due to their hydrophilic nature as well as better biocompatibility.[Citation30–33] This is because certain glycans have ability to regulate cellular functions due to their flexibility for spatial distribution in organization of glyco moieties and functional multivalent nature of glycoconjugates.[Citation34,35] The coatings of glycans on metals and poly(ethylene oxide) (PEO) surfaces have been for eternity challenging to functionalise these with glycans and very limited studied were available in the literature to describe the effects of glycopolymers architectures on cellular functions of bone cells. The biological stimuli such as using bone morphogenetic proteins (BMP) have the potential to obtain new bone formation in-vivo which could be very useful for the repair of bone injuries and bone defects. However, the major challenge remains in the development of the appropriate delivery system which allows an optimal local delivery of the BMP. Previous in-vivo studies suggest that type I collagen may be preferred as a delivery system for biological stimuli;[Citation36,37] however, the biological material carries potential risks for disease-transfer and immune reaction when used in a clinical setting.[Citation38–40] To overcome with these limitations, Saito et al. [Citation41] had developed a synthetic biodegradable polymer – poly(D,L-lactic acid)–p-dioxanone–poly(ethylene glycol) block copolymer. They reported that the polymer exhibits promising degradation characteristics as a delivery system for biological stimuli and were of good biocompatibility. Similarly, a range of novel bioactive materials with different mechanical properties were developed by Kokubo et al. [Citation42]. Recently, Humber et al. [Citation43] had reported bone healing with an in situ-formed bio-resorbable poly(ethylene glycol) (PEG) hydrogel membrane in rabbit calvarial defects. Considering above findings, we have attached glycopolymer functionalities with PEO block where the glycopolymer can act as a chemical stimuli for better cell growth whereas PEO helpful in the control delivery. It has been hypothesized that the functionalization of implant surfaces with glycopolymer functional moieties may initiate biologically active response with cells/tissues similar to the natural systems.[Citation34,35] As per literature, the biocompatibile PEO-based glycopolymers were used in-vivo self-regulated insulin delivery system [Citation44] and moreover the PEG-based glycopolymers found to be superior lyophilization stability.[Citation45] Thus, the synthesis of glycopolymers having PEO/PEG segment may provide new opportunity to functionalise implant materials for biomedical and implant uses.
Previously, controlled macromolecular architectures of glycopolymers were frequently reported by controlled radical processes from a wide range of functional monomers.[Citation46–51] Narain and Armes reported that PEO-based 2-gluconamidoethyl methacrylate di-block copolymers synthesis by atom-transfer radical polymerization (ATRP) method in protic media.[Citation52] Eventually, the authors studied reversible micellar self-assembly of PEO-based block copolymers in aqueous solution as a function of pH and temperature.[Citation53] The reversible formation of aggregates from a dilute solution of ATRP generated PEO-based di-block glycopolymers in the presence of Con A and glucose give an indication as useful in drug delivery system.[Citation54] The di-block copolymers with poly(2-glucosyloxyethyl methacrylate) as one segment and poly(diethyleneglyco methacrylate) as a second block were synthesized to produce a mimic of eukaryotic cell surfaces, which can recognize biological species and these glycopolymer can bind receptors through polyvalency.[Citation55] Amphiphilic poly(ε-caprolactone)-b-poly[(methacrylate-graft-poly(ethylene oxide))-co-6-O-methacryloyl-d-galactopyranose] copolymers have been prepared by combining coordination-insertion ring-opening polymerization, ATRP and followed by deprotection process. The resulted copolymers exhibited micellar aggregates in aqueous media by self-assembly nature.[Citation56] Here we have employed RAFT process to synthesize PEO segmented multivalent glycopolymers containing three different glucose pendant spacer lengths to the macromolecular backbone. Further, we report the pendant alkyl chain lengths of glucose functional moieties response on MC3T3 cells at various concentrations of di-block copolymers in terms of cell adhesion, viability, and proliferation.
Experimental details
Materials
D-glucose anhydrous (Fisher Scientific Pvt. Ltd), acetic anhydride and sodium acetate (Sisco Research Laboratories Pvt. Ltd.), benzylamine (Finar Chemicals Limited), methyl ethyl ketone (MEK), acryloylchloride, triethylamine (Et3N), CH3ONa and azobisisobutyronitrile (AIBN) (Avra Synthesis Pvt. Ltd.), 1,4-butane diol, 1,6-hexane diol, boron trifluoride diethyl etherate (BF3·Et2O,) and PEO methyl ether (4-cyano-4-pentanoate dodecyl trithiocarbonate) average Mn: 2400, methanol (Sigma–Aldrich) and molecular sieves 4 Å (S d fine-chem limited), 1,4-dioxane (Spectro Chem Pvt Ltd) and CHCl3 (R F C L ltd) were used as received. Dichloromethane (DCM) (Finar Chemicals Ltd) was dried over calcium hydride by stirring over night and distilled. The cell culture medium (CM) was prepared using α-MEM medium, supplemented with 10% FBS and 100 U/mL penicillin, 50 μg/mL streptomycin, and 50 μg/mL gentamycin. The stock solutions of different concentrations of all three PEO-based di-block glycopolymers were prepared in phosphate-buffered saline (PBS).
Synthesis of glycomonomers
Acrylate-based glycomonomers having different aliphatic spacer lengths were synthesized by standard procedures. In the first step, 1,2,3,4,6-penta-O-acetyl-d-glucopyranoside (PAGP) was prepared by reacting D-glucose with sodium acetate in the presence of acetic anhydride. The PAGP treated with benzyl amine in THF to yield 2,3,4,6-penta-O-acetyl-d-glucopyranoside (TAGP).[Citation57] The 6′-hydroxyhexyl-2,3,4,6-tetra-O-acetyl-d-glucopyranoside (HHTAGP) and 4′-hydroxybutyl-2,3,4,6-tetra-O-acetyl-d-glucopyranoside [Citation58] was obtained by treating the PAGP with 1,4-butanediol/1,6 hexanediol in the presence of BF3·Et2O in DCM. The TAGP, ABTAGP, and HHTAGP were further reacted with acryl chloride in the presence of triethylamine in MEK [Citation59] to obtain the acryl-2,3,4,6-tetra-O-acetyl-d-glucopyranoside (ATAGP), 4′-(acryloxy)butyl-2,3,4,6-tetra-O-acetyl-d-glucopyranoside (ABTAGP) and 6′-(acryloxy)hexyl-2,3,4,6-tetra-O-acetyl-d-glucopyranoside (AHTAGP).
Synthesis of PEO-based di-block glycopolymers
Preparation of poly(ethylene oxide)-b-poly(acryl-2,3,4,6-tetra-O-acetyl-d-glucopyranoside) [PEO-b-P(ATAGP)] (1a)
A mixture of acryl-2,3,4,6-tetra-O-acetyl-d-glucopyranoside (3 g, 0.0075 mol), AIBN (33 mg, 0.0002 mol), and a macro RAFT agent, PEO methyl ether (4-cyano-4-pentanoate dodecyl trithiocarbonate) (716 mg, 0.0003 mol) in 15 ml of 1,4-dioxane was deoxygenated by three times of freeze-pump-thaw cycles before sealing the flask. Then, the flask was heated to 80 °C under stirring. It was allowed for 30 h. After that, the polymerization was stopped by the addition of 0.5 ml of methanol and exposed to air. The polymer was isolated by twofold precipitation in cold methanol and one time in 30% mixture of ethyl acetate in hexane. Yield: 53%. 1H NMR (CDCl3, 300 MHz): δ 6.52–6.19, 5.89–5.60, 5.54–5.36, 5.34–5.24, 5.21–4.99, 4.52–4.22, 4.21–4.01, 3.99–3.80, 3.70–3.58, 3.54–3.41, 2.80–2.28, 2.26–1.89, 1.87–1.36 and 1.32–0.97. 13C NMR (CDCl3, 75 MHz): δ 19.16–21.33, 30.65, 32.95–36.73, 38.49–42.68, 60.63, 61.65, 67.51, 69.02, 69.42, 70.36, 72.51, 89.11, 91.65, 169.29, 169.92 and 170.47. FT-IR (KBr, cm−1): ν 2963 (–CH–), ν 1755 (–O–C=O–), ν 1631 (–C=S–), ν 1438, 1372 (–O–C–, ester) and ν 1224 (–C–O–C–).
Preparation of PEO-b-poly(4’-(acryloxy)butyl-2,3,4,6-tetra-O-acetyl-d-glucopyranoside) [PEO-b-P(ABTAGP)] (1b)
A mixture of 4′-(acryloxy)butyl-2,3,4,6-tetra-O-acetyl-d-glucopyranoside (3.5 g, 0.0073 mol), AIBN (164 mg, 0.0001 mol) and PEO methyl ether (4-cyano-4-pentanoate dodecyl trithiocarbonate) (480 mg, 0.0002 mol) in 20 ml of 1,4-dioxane was deoxygenated by three times of freeze-pump-thaw cycles before sealing the flask. Followed by the flask was heated to 80 °C under stirring, and after 30 h the above procedure was followed to obtain PEO-b-P(ABTAGP). Yield: 30%.1H NMR (CDCl3, 300 MHz): δ 5.80–5.56, 5.51–4.63, 4.42–4.16, 4.15–3.92, 3.91–3.78, 3.76–3.68, 3.67–3.39, 3.38–3.18, 2.61–2.25, 2.21–1.77, 1.75–1.46, 1.43–1.08 and 0.97–0.76. 13C NMR (CDCl3, 75 MHz): δ 14.09, 20.49, 22.59, 25.72, 25.46, 28.47, 29.12, 29.51, 31.74, 40.11, 42.46, 59.06, 61.42, 67.17, 67.95, 68.35, 69.40, 69.92, 70.44, 71.74, 72.53, 90.18, 169.41, 169.94 and 170.46. FT-IR (KBr, cm−1): ν 2962 (–CH–), ν 1755 (–O–C=O–), ν 1634 (–C=S–), ν 1439, 1372 (–O–C–, ester) and ν 1226 (–C–O–C–).
Preparation of PEO-b-poly(6′-(acryloxy)hexyl-2,3,4,6-tetra-O-acetyl-d-glucopyranoside) [PEO-b-P(AHTAGP)] (1c)
A mixture of 6′-(acryloxy)hexyl-2,3,4,6-tetra-O-acetyl-d-glucopyranoside (1 g, 0.002 mol), AIBN (8.8 mg, 0.00005 mol) and poly(ethylene glycol) methyl ether (4-cyano-4-pentanoate dodecyl trithiocarbonate) (191 mg, 0.00008 mol) in 20 ml of 1,4-dioxane was deoxygenated by three times of freeze–pump–thaw cycles. Subsequently, the polymerization pot was sealed and heated to 80 °C under stirring. After 30 h, the PEO-b-P(AHTAGP) was obtained by following above procedure. Yield: 52%. 1H NMR (CDCl3, 300 MHz): δ 6.45–6.20, 5.93–5.57, 5.54–5.37, 5.37–5.22, 5.22–4.91, 4.57–4.20, 4.19–4.01, 4.00–3.81, 3.80–3.57, 3.56–3.52, 3.52–3.46, 3.18–3.04, 2.81–2.30, 2.26–1.88, 1.87–1.49 and 1.48–1.08. 13C NMR (CDCl3, 75 MHz): δ 8.42, 20.37, 25.39, 28.32, 29.24, 30.66, 31.70–35.66, 37.82, 39.0–41.90, 45.61, 58.81, 61.47, 64.18, 67.45, 69.57, 70.31, 72.4, 73.2, 91.97, 169.20, 169.84 and 170.37. FT-IR (KBr, cm−1): ν 2945 (–CH–), ν 1756 (–O–C=O–), ν 1633 (–C=S–), ν 1439, 1372 (–O–C–, ester) and ν 1226 (–C–O–C–).
Deacetylation of pendant 2,3,4,6-tetra-O-acetyl-d-glucopyranoside moieties
All the three di-block copolymers pendant 2,3,4,6-tetra-O-acetyl-d-glucopyranoside moieties were deacetylated. A typical procedure is as follows.
The synthesized di-block copolymer (200 mg) was dissolved in anhydrous methanol and chloroform (2:1) (total volume: 10 ml), and stirred under a nitrogen atmosphere. A solution of NaOMe (101 mg) in anhydrous methanol (2 ml) was added through a syringe and the reaction mixture was stirred for 90 min. Then solvent was removed by vacuum evaporation and neutralized with 2 M HCl solution by stirring over night. After that, the solution was passed through the basic alumina to neutralize the solution. Excess water was removed by lyophilization and the di-block copolymer was recovered as a white powder by precipitation in excess acetone.
Poly(ethylene oxide)-b-poly(acryl-d-glucopyranoside) (PEO-b-PAG) (2a): FT-IR (KBr, cm−1): ν 3425 (–OH–), 2929 (–CH–), 1730 (–O–C=O–), 2200 (–CN), 1592 (–C=S–) and 1059 (C–O–C).
Poly(ethylene oxide)-b-poly(4′-(acryloxy)butyl-d-glucopyranoside (PEO-b-PABG) (2b): FT-IR (KBr, cm−1): ν 3420 (–OH–), 2927 (–CH–), 1736 (–O–C=O–), 2200 (–CN), 1629 (–C=S–) and 1053 (C–O–C).
Poly(ethylene oxide)-b-poly(6′-(acryloxy)hexyl-d-glucopyranoside) (PEO-b-PAHG) (2c): FT-IR (KBr, cm−1): ν 3424 (–OH–), 2926 (–CH–), 1738 (–O–C=O–), 2200 (–CN), 1594 (–C=S–) and 1079 (C–O–C).
In-vitro cell assays
Cell culture and statistical analysis
The MC3T3 were cultured in α-MEM at 37 °C in 5% CO2. Different concentrations of glycopolymer were added by adding 4 μL of stock solution of different desired concentration in 96 μL of CM for each set of experiments and then 3 × 103 cells were plated well in each of the plate. Statistical analysis for in-vitro cell assay was done, based on three experiments for each condition in which minimum three samples were used. Microsoft excel software was used for calculating mean and standard deviation values for all assays.
Cell adhesion and viability assays
These assays were performed by plating similar number of cells and after 4 and 24 h non- adherent cells were washed off and 10 μL of 2 mg/mL MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was added to the remaining cells of each condition followed by incubation at 37 °C and 5% CO2 for cell adhesion and viability assessment, respectively. This reaction was stopped after removing CM and by adding dimethylsulfoxide and optical density was read at 595 nM in ELISA reader.
Cell proliferation assay
The assay was performed by plating similar number of cells of all conditions in triplicates and incubated at dissimilar time periods namely, 24, 48, and 72 h. At the specified time periods, cell number at each concentration was calculated by MTT assay as described above.
Immunofluorescence staining
Cells were plated in 6-well plate and incubated for up to 24 h at various concentration of glycopolymers. The cells were fixed with 4% formaldehyde (Sigma, USA) for 5–6 min. Cells were washed with PBS for three times and permeabilization was done with 0.1% Triton X-100 for 5–6 min following three times washing with PBS. About 3% bovine serum albumin (BSA) was used for blocking for 1 h, and finally cells were washed with PBS. Integrin α5 and talin co-staining was performed as follow, after 24 h cells on the above substrates were fixed in 4% paraformaldehyde for 10 min at room temperature, permeabilized with 0.1% Triton-X-100 for 10 min and blocked with 3% BSA (Bovine Serum Albumin, Sigma) for 30 min. The cells were treated with rabbit anti-mouse integrin α5 antibody at 1:200 (Santa Cruz Biotechnology, Texas, USA) for 1 h at room temperature followed by 1 h incubation with goat anti-rabbit FITC (Fluorescein isothiocyanate) at 1:200 (GeneI, Bangalore, India). Same procedure and conditions repeated for talin staining with mouse anti-mouse talin antibody followed by a 1 h incubation with goat anti-mouse TRITC in the dark at room temperature. Finally, cells were washed once with PBS and mounted using fluoroshield 4′,6-diamidino-2-phenylindole (DAPI) (Sigma, USA). Cells were imaged with fluorescent microscope (Axio Imager.Z1, Carl Zeiss, Jena Germany). Images of cells were captured using a 40× or 63× objectives separately in an AxioVision imaging system (Zeiss, Germany).
Characterization
The FT-IR (Thermo Nicolet Nexus 670 spectrometer) spectra were recorded at a resolution of 4 cm−1 using KBr optics at room temperature and 32 scans were signal averaged. Mass spectra were recorded on FINNGAN LCQ Advance Max in methanol. The proton nuclear magnetic resonance (1H NMR) and carbon nuclear magnetic resonance (13C NMR) spectra were measured at room temperature by using AVANCE-300 or INOVA-500 spectrometer in CDCl3 or D2O (Aldrich) depends on the solubility of the di-block copolymers with tetramethylsilane as a reference. Gel permeation chromatography (GPC) was performed by using a Shimadzu system built-in with one (300 × 7.5 mm) PLgel 5 μM 10E3 Å column (VARIAN) and evaporative light-scattering (PL-ELS) detector (Polymer Laboratories). The eluent was DMF with a flow rate of 0.5 ml min−1 at 30 °C. Calibration for detector response was gained using single narrow PS standards (molecular weights: 550, 1480, 3950, 10,680, and 31,420 from Polymer Labs). 1 mg of polymer was dissolved in 1 ml of DMF. The values of Mn and Mw/Mn were determined using LC Solution, Windows software. CO2 incubator was purchased from Thermo Scientific, USA. Spectrophotometric measurements of adhesion and proliferation were taken from Molecular Devices-Spectra Max-190, USA.
Results and discussion
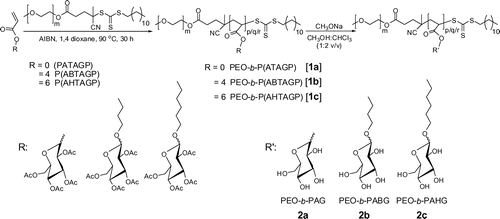
The PEO based di-block copolymers with glycoacrylates, ATAGP, ABTAGP, and AHTAGP were prepared by RAFT process using PEO-methyl ether (4-cyano-4-pentanoate dodecyl trithiocarbonate) as macro RAFT agent with azobisisobutyronitrile as a radical initiator. The resulted PEO-b-P(ATAGP) (1a), PEO-b-P(ABTAGP) (1b), and PEO-b-P(AHTAGP) (1c) di-block copolymers pendant 2,3,4,6-tetra-O-acetyl-d-glucopyranoside units were deacetylated in the presence of sodiummethoxide/chloroform/methanol mixture to obtain the PEO-b-PAG (2a), PEO-b-PABG (2b), and PEO-b-PAHG (2c), respectively. The structures of the copolymers before and after deacetylation are given in Scheme .
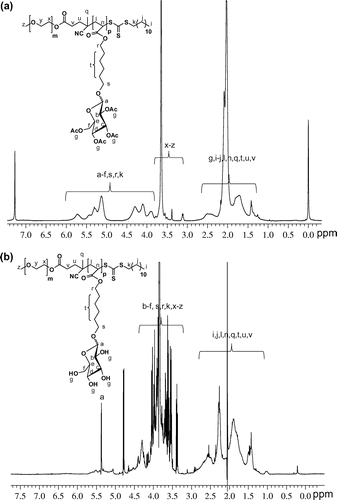
Before and after deacetylation of the di-block copolymers showed monomodal SEC curves and the molar masses with polydispersities (Mw/Mn) are summarized in Table . The calculated molecular weights of the copolymers from 1H NMR spectra and the SEC values had good agreement with each other except the 1c and 2a copolymers. The deacetylated di-block glycopolymers were soluble in water which were insoluble with having acetylated moieties. The acrylate based acetylated block glucose pendants 2,3,4,6-tetra-O-acetyl signals integrations showed at δ 1.8–2.3 ppm in 1H NMR spectra as shown in Figure (a). The pendant acetyl peaks disappeared after deacetylation of the di-block copolymers and new signals corresponding to –OH groups of the glucose moieties [Citation55] were emerged at δ 3.8–4.0 ppm (Figure (b)). The FT-IR spectra of copolymers showed a characteristic strong absorptions for C=O stretching centered at about 1756 cm−1 whereas the deacetylated copolymers showed the C=O stretching peaks in the range of 1730–1736 cm−1 with lower intensities (ESI Figures S5 and S6). The –OH signals of the acetylated di-block copolymers showed characteristic strong absorptions centred at 3425 cm−1.
Table 1. Acetylated and deacetylated di-block copolymers characteristics.
Figure 1. 1H -NMR spectra of (a) 1c in CDCl3 and (b) 2c in D2O.
Recently, we have reported multivalent glycopolymers containing three different pendant spacer lengths between polymer back bone and glucose moiety.[Citation60] The biological responses of these homoglycopolymers, in terms of cytotoxicity of glucose moieties at various spacer length and concentrations, were investigated by employing in-vitro osteoblast cells. Osteoblast cells adhesion, viability, and proliferation response with synthesized homoglycopolymers revealed that the homoglycopolymer concentration tolerance limit was higher with an increase in the pendant spacer length of glucose moiety of the homoglycopolymer. The chemical structure played a vital role in homopolymers. Owing to that reason, in the present study, we employed PEO-based di-block glycopolymers containing three different pendant spacer length glucose moieties of glycopolymer chains to understand cyototoxicity of in-vitro osteoblast cells.
Figure (A) shows cell adhesion % on tissue culture plastic (TCP) surfaces without the use of glycopolymers. The cell adhesion % at various concentrations of 2a, 2b, and 2c in the cell culture media is shown in Figure (B). The use of 10 and 100 μM of 2b and 2c showed the highest response of osteoblast cells adhesion. However, a further increase in the concentration (1000 μM) of 2a, 2b, and 2c showed rapid decrease in the osteoblast cells adhesion. At the highest concentration (1000 μM) of glycopolymers used in the experiment, the 2b showed better cell adhesion as compared to the other two glycopolymers. Conversely, the decrease in di-block copolymers concentrations <10 μM showed a decrease in adhesion of the cells and it remains less than the control conditions (without the use of copolymer).
Figure 2. Osteoblast cells (A) adhesion after 4 h and (B) viability after 24 h at different concentrations of (a) control, (b) 10 nM, (c) 100 nM, (d) 1 μM, (e) 10 μM, (f) 100 μM, and (g) 1000 μM concentrations of di-block glycopolymers.
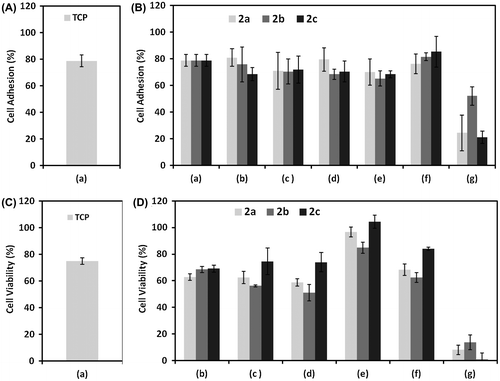
The cell viability % at various concentrations of copolymers is shown in Figure (C) and (D) and the relative change in the % cell viability compared with control conditions on TCP. The use of 10 μM of all copolymers exhibited highest % cell viability. There were no significant change in cell viability % was occurred at the use of different concentrations (10 nM to 100 μM) of 2c as compared to the cells viability obtained for control conditions on TCP. The use of low concentrations of 2a and 2b showed relative decrease in cell viability %. Similarly, a further increase (>100 μM) in the concentration had showed steep decrease in cell viability % for all the copolymers. Relatively, the % cell viability was better for 2b at 1000 μM concentration as compared to the other two glycopolymers.
To assess the response of cell spreading in the presence of glycopolymer, we have obtained microscopic images of cells stained with integrin α5 and talin at various concentrations of 2a, 2b, and 2c copolymers and results are shown in Figure . We have chosen integrin α5 staining for assessing the spreading of cells because it exhibit functional role of the cytoplasmic domain of the alpha subunit of the integrin which play an important role in determining cell surface interaction. We have co-localized integrin α5 with talin to demonstrate the focal contact points with glycopolymers and without glycopolymers treatment as experimental control (ESI, Figure S9).
Figure 3. Fluorescent microscopy images of co-stained, integrin α5, talin and DAPI, and merged for assessing the expression of cytoskeleton protein at (a) 10 nM, (b) 10 μM, and (c) 100 μM concentrations of di-block copolymers.
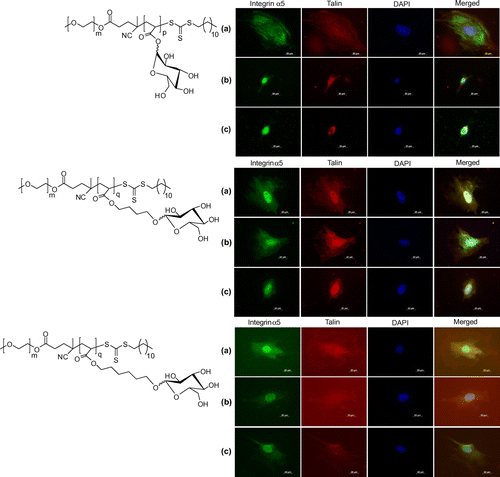
Talin and integrin α5 immunostaining were performed at 10 nM, 10, and 100 μM for 2a, 2b, and 2c and cell cytoskeletal organization was compared with relative change in the concentrations of each glycopolymer. From the cell staining images, it was clearly indicative that cells were spreading well in the presence of all glycopolymers at 10 nM of their concentration. A further increase in the concentration of 2a at 10 and 100 μM had showed significant change in the cell surface morphologies, despite having good cell adhesion and viability as compared to the control conditions. The use of 10 and 100 μM concentrations of 2c did not exhibit much change in the cell spreading. The use of 100 μM concentrations of 2b had showed significant decrease in the cell size and focal contact points as compared to the cell spreading at control conditions.
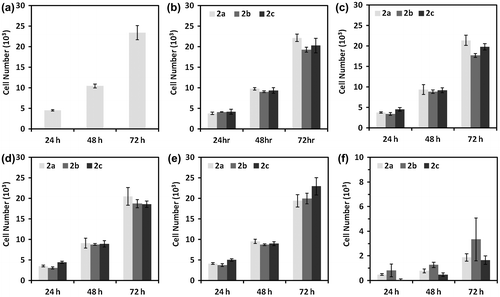
Osteoblast cells proliferation was estimated at 24, 48, and 72 h after cell platting at various concentrations (0–1000 μM) of copolymers and the results are shown in Figure . At lower concentrations, from 0 to 100 μM, the cells proliferation did not exhibit any significant change with change in pendant alkyl chain length of glucose moiety of glycopolymer segment in di-block copolymer. The cells proliferation was reduced rapidly with a further increase in concentration of copolymers and most of the cells had lost their viability at the use of 1000 μM concentration. In this study, we observed that osteoblast cell proliferation in the presence of PEO segmented glycopolymers was not significantly changed with the change in pendant spacer length of the glucose moiety in the acrylate glycopolymer segment. Previously, we had demonstrated that the RAFT generated acrylate glycopolymers without PEO segment had showed that cell proliferation tolerance was extended by increasing the pendant spacer length of the glucose moiety in glycopolymer segment.[Citation60] In the present study, the observed findings demonstrate that the concentration of functional moiety of the glycopolymer play an important role in determining the biocompatibility of PEO-segmented acrylate glycopolymers whereas the cell spreading and morphology depends on the spacer length of the glucose moiety in di-block copolymers.
Figure 4. Osteoblast cells proliferation at (a) control, (b) 10 nM, (c) 100 nM, (d) 1 μM, (e) 100 μM, and (f) 1000 μM concentrations of di-block copolymers at 24, 48, and 72 h after cell platting.
Scheme 1. Schematic representation of the syntheses of PEO based di-block glycopolymers (0, 4, and 6 indicates spacer length) via the RAFT process and deacetylation of the pendant 2,3,4,6-tetra-O-acetyl-d-glucopyranoside.
Conclusions
We have synthesised di-block copolymers containing PEO and glycopolymers with three different pendant spacer lengths of glucopyranoside by deacetylation of pendant acetyl moieties which were prepared by RAFT process. The effect of PEO segmented di-block copolymers having various spacer lengths of glucose moiety on osteoblast cells adhesion, viability, and proliferation was investigated. It was demonstrated that the all three di-block glycopolymers has showed very good biocompatibility up to the use of 100 μM of their concentrations. The cell spreading and morphology depends on the use of concentration and spacer length of the glucose moiety in di-block copolymers. The synthesis of di-block copolymer of PEO and glycopolymer may provide the possibility of attachment of various types of glycan moieties on PEO base implant materials for preparing biologically active functional implant surfaces.
Supplementary material
The supplementary material for this paper is available online at http://dx.doi.10.1080/15685551.2015.1092009.
Supplementary_data.pdf
Download PDF (1.6 MB)Acknowledgment
We thank CSIR-CCMB microscopy facility. MT, VS and GK thank CSIR and CSIR network project for providing fellowships.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
AVSS and MD thank the Council for Scientific and Industrial Research, India for financial support in network projects DENOVA CSC-0205 and BSC-0112, respectively.
References
- Kurtz S, Ong K, Lau E, et al. Projections of primary and revision hip and knee arthroplasty in the United States from 2005–2030. J. Bone Joint Surg. Am. 2007;89:780–785.10.2106/JBJS.F.00222
- Trampuz A, Widmer AF. Infections associated with orthopaedic implants. Curr. Opin. Infect. Dis. 2006;19:349–356.10.1097/01.qco.0000235161.85925.e8
- Hubbell JA. Biomaterials in tissue engineering. Biotechnology. 1995;13:565–576.10.1038/nbt0695-565
- Nathaniel H, Mooney DJ. Review article inspiration and application in the evolution of biomaterials. Nature. 2009;462:426–432.
- Kohn J. New approaches to biomaterials design. Nat. Mater. 2004;3:745–747.10.1038/nmat1249
- Brahatheeswaran D, Yasuhiko Y, Toru M, et al. Polymeric Scaffolds in tissue engineering application: a review. Int. J. Polym. Sci. 2011; Article ID 290602:1–19.
- Rajesh V, Dhirendra SK. Nanofibers and their applications in tissue engineering. Int. J. Nanomed. 2006;1:15–30.
- Lutolf MP, Weber FE, Schmoekel HG, et al. Repair of bone defects using synthetic mimetics of collagenous extracellular matrices. Nat. Biotechnol. 2003;21:513–518.10.1038/nbt818
- You Z, Cao H, Gao J, et al. A functionalizable polyester with free hydroxyl groups and tunable physiochemical and biological properties. Biomaterials. 2010;31:3129–3138.10.1016/j.biomaterials.2010.01.023
- Dhayal M, Kapoor R, Sistla PG, et al. Strategies to prepare TiO2 thin films, doped with transition metal ions, that exhibit specific physicochemical properties to support osteoblast cell adhesion and proliferation. Mater. Sci. Eng. C. 2014;37:99–107.10.1016/j.msec.2013.12.035
- Hutmacher DW. Scaffold design and fabrication technologies for engineering tissues-state of the art and future perspectives. J. Biomater. Sci. Polym. Ed. 2001;12:107–124.10.1163/156856201744489
- Hutmacher DW. Scaffolds in tissue engineering bone and cartilage. Biomaterials. 2000;21:2529–2543.10.1016/S0142-9612(00)00121-6
- Okamoto M, John B. Synthetic biopolymer nanocomposites for tissue engineering scaffolds. Prog. Polym. Sci. 2013;38:1487–1503.10.1016/j.progpolymsci.2013.06.001
- Liu Y, Lim J, Teoh S-W. Review: development of clinically relevant scaffolds for vascularised bone tissue engineering. Biotechnol. Adv. 2013;31:688–705.10.1016/j.biotechadv.2012.10.003
- Geetha M, Singh AK, Asokamani R, et al. Ti based biomaterials, the ultimate choice for orthopaedic implants – a review. Prog. Mater Sci. 2009;54:397–425.10.1016/j.pmatsci.2008.06.004
- Long M, Rack HJ. Titanium alloys in total joint replacement – a materials science perspective. Biomaterials. 1998;19:1621–1639.10.1016/S0142-9612(97)00146-4
- Ben-Nissan B, Choi AH, Bendavid A. Mechanical properties of inorganic biomedical thin films and their corresponding testing methods. Surf. Coat. Technol. 2013;233:39–48.10.1016/j.surfcoat.2012.11.020
- Chen W, Villa-Diaz LG, Sun Y, et al. Nanotopography influences adhesion, spreading, and self-renewal of human embryonic stem cells. ACS Nano. 2012;6:4094–4103.10.1021/nn3004923
- Kim BC, Moraes C, Huang J, et al. Fracture-based micro-and nanofabrication for biological applications. Biomater. Sci. 2014;2:288–296.10.1039/c3bm60276a
- Curtis ASG, Dalby M, Gadegaard N. Cell signalling arising from nanotopography: implications for nanomedical devices. Nanomedicine. 2006;1:67–72.
- Mohammed JS, DeCoster MA. McShane MJ, Micropatterning of nanoengineered surfaces to study neuronal cell attachment in vitro. Biomacromolecules. 2004;5:1745–1755.
- Higuchi A, Ling Q-D, Chang Y, et al. Physical cues of biomaterials guide stem cell differentiation fate. Chem. Rev. 2013;113:3297–3328.10.1021/cr300426x
- Mahmoudi M, Bonakdar S, Shokrgozar MA, et al. Cell-imprinted substrates direct the fate of stem cells. ACS Nano. 2013;7:8379–8384.10.1021/nn403844q
- Dai L, Mau AWH. Surface and interface control of polymeric biomaterials, conjugated polymers, and cabon nanotubes. J. Phys. Chem. B. 2000;104:1891–1915.10.1021/jp9926793
- Vijay YK, Dhayal M, Awasthi K, et al. Surface modification and synthesis of polymeric membrane for energy and biological applications. J. Biomed. Nanotechnol. 2006;2:144–151.10.1166/jbn.2006.026
- Zhao Y, Wong SM, Wong HW, et al. Effects of carbon and nitrogen plasma immersion ion implantation on in vitro and in vivo biocompatibility of titanium alloy. ACS Appl. Mater. Interfaces. 2013;5:1510–1516.10.1021/am302961h
- Desmet T, Morent R, Geyter ND, et al. Nonthermal plasma technology as a versatile strategy for polymeric biomaterials surface modification: a review. Biomacromolecules. 2009;10:2351–2378.10.1021/bm900186s
- Dhayal M, Cho SI, Moon JY, et al. S180 cell growth on low ion energy plasma treated TiO2 thin films. Appl. Surf. Sci. 2008;254:3331–3338.10.1016/j.apsusc.2007.11.026
- Loty C, Sautier JM, Tan MT, et al. Bioactive glass stimulates in-vitro osteoblast differentiation and creates a favorable template for bone tissue formation. J. Bone Miner. Res. 2001;16:231–239.10.1359/jbmr.2001.16.2.231
- Kiessling LL, Grim JC. Glycopolymer probes of signal transduction. Chem. Soc. Rev. 2013;42:4476–4491.10.1039/c3cs60097a
- Ladmiral V, Melia E, Haddleton DM. Synthetic glycopolymers: an overview. Eur. Polym. J. 2004;40:431–449.10.1016/j.eurpolymj.2003.10.019
- Ambrosi M, Cameron NR, Davis BG, et al. Investigation of the interaction between peanut agglutinin and synthetic glycopolymeric multivalent ligands. Org. Biomol. Chem. 2005;3:1476–1480.10.1039/b411555b
- Spain SG, Gibson MJ, Cameron NR. Recent advances in the synthesis of well-defined glycopolymers. J. Polym. Sci., Part A: Polym. Chem. 2007;45:2059–2072.10.1002/(ISSN)1099-0518
- Dam TK, Brewer FC. Maintenance of cell surface glycan density by lectin–glycan interactions: a homeostatic and innate immune regulatory mechanism. Glycobiology. 2010;20:1061–1064.10.1093/glycob/cwq084
- Varki A. Biological roles of oligosaccharides: all of the theories are correct. Glycobiology. 1993;3:97–130.10.1093/glycob/3.2.97
- Takaoka K, Koezuka M, Nakahara H. Telopeptide-depleted bovine skin collagen as a carrier for bone morphogenetic protein. J. Orthop. Res. 1991;9:902–907.10.1002/(ISSN)1554-527X
- Rodeo SA, Suzuki K, Deng X, et al. Use of recombinant bone morphogenetic protein-2 to enhance tendon healing in a bone tunnel. Am. J. Sports Med. 1999;27:476–488.
- DeLustro F, Dasch J, Keefe J, et al. Immune responses to allogeneic and xenogeneic implants of collagen and collagen derivatives. Clin. Orthop. 1990;260:263–279.
- Butler D, Wadman M, Lehrman S, et al. Last chance to stop and think on risks of xenotransplants. Nature. 1998;391:320–324.
- Bach FH, Fishman JA, Daniels N, et al. Uncertainty in xenotransplantation: Individual benefit versus collective risk. Nat. Med. 1998;4:141–144.10.1038/nm0298-141
- Saito N, Okada T, Horiuchi H, et al. A biodegradable polymer as a cytokine delivery system for inducing bone formation. Nat. Biotechnol. 2001;19:332–335.10.1038/86715
- Kokubo T, Kim H-M, Kawashita M. Novel bioactive materials with different mechanical properties. Biomaterials. 2003;24:2161–2175.10.1016/S0142-9612(03)00044-9
- Humber CC, Sándor GK, Davis JM, et al. Bone healing with an in situ-formed bioresorbable polyethylene glycol hydrogel membrane in rabbit calvarial defects. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2010;109:372–384.10.1016/j.tripleo.2009.10.008
- Yang H, Sun X, Liu G, et al. Glucose-responsive complex micells for self-regulated release of insulin under physiological conditions. Soft Matter. 2013;9:8589–8599.10.1039/c3sm51538a
- Mancini RJ, Lee J, Maynard HD. Trehalose glycopolymers for stabilization of protein conjugates to environmental stressors. J. Am. Chem. Soc. 2012;134:8474–8479.10.1021/ja2120234
- Ghadban A, Albertin L. Synthesis of glycopolymer architectures by reversible-deactivation radical polymerization. Polymers. 2013;5:431–526.10.3390/polym5020431
- Miura Y. Design and synthesis of well-defined glycopolymers for the control of biological functionalities. Polym. J. 2012;44:679–689.10.1038/pj.2012.4
- Lowe AB, Sumerlin BS, McCormick CL. The direct polymerization of 2-methacryloxyethyl glucoside via aqueous reversible addition-fragmentation chain transfer (RAFT) polymerization. Polymer. 2003;44:6761–6765.10.1016/j.polymer.2003.08.039
- Cameron NR, Spain SG, Kingham JA, et al. Synthesis of well-defined glycopolymers and some studies of their aqueous solution behaviour. Faraday Discuss. 2008;139:359–368.10.1039/b717177c
- Wang Y, Zhang X, Mu J, et al. Synthesis and pH/sugar/salt-sensitivity study of boronate crosslinked glycopolymer nanoparticles. New J. Chem. 2013;37:796–803.10.1039/c2nj40998d
- Zhang Q, Haddleton DM. Synthetic glycopolymers: some recent developments. Adv. Polym. Sci. 2013;262:39–59.10.1007/978-3-319-03719-6
- Narain R, Armes SP. Synthesis of low polydispersity, controlled-structure sugar methacrylate polymers under mild conditions without protecting group chemistry. Chem. Commun. 2002;23:2776–2777.
- Narain R, Armes SP. Synthesis and aqueous solution properties of novel sugar methacrylate-based homopolymers and block copolymers. Biomacromolecules. 2003;4:1746–1758.10.1021/bm034166e
- You L-C, Lu F-Z, Li Z-C, et al. Gluose-sensitive aggregates formed by poly(ethylene oxide)-block-poly(2-glucosyl-oxyethyl acrylate) with concanavalin A in dilute aqueous medium. Macromolecules. 2003;36:1–4.10.1021/ma025641o
- Pasparakis G, Alexander C. Sweet talking double hydrophilic block copolymer vesicles. Angew. Chem. Int. Ed. 2008;47:4847–4850.10.1002/(ISSN)1521-3773
- Suriano F, Coulembier O, Degée P, et al. Carbohydrate-based amphiphilic diblock copolymers: synthesis, characterization, and aqueous properties. J. Polym. Sci., Part A: Polym. Chem. 2008;46:3662–3672.10.1002/(ISSN)1099-0518
- Sim M, Kondo H, Wong CH. Synthesis of dibenzyl glycosyl phosphites using dibenzyl N,N-diethylphosphoramidite as phosphitylating reagent: an effective route to glycosyl phosphates, nucleotides, and glycosides. J. Am. Chem. Soc. 1993;115:2260–2267.10.1021/ja00059a023
- Woodcock JW, Jiang X, Wright RAE, et al. Enzyme-induced formation of thermoreversible micellar gels from aqueous solutions of multiresponsive hydrophilic ABA triblock copolymers. Macromolecules. 2011;44:5764–5775.10.1021/ma200991d
- Petrovica Z, Konstantinovic S, Spasojevic A. BF3 etherate-induced formation of C7-C16-alkyl-β-d-glucopyranosides. Indian J. Chem. Sect. B. 2004:132–134.
- Trinadh M, Kannan G, Rajasekhar T, et al. Synthesis of glycopolymers at various pendant spacer lengths of glucose moiety and their effects on adhesion, viability and proliferation of osteoblast cells. RSC Adv. 2014;4:37400–37410.10.1039/C4RA05436A