
Abstract
New biosourced unprotected diols were prepared by acylation reaction of aminoalcohol based on 1,4:3,6-dianhydrosorbitol (Isosorbide Is) with several aliphatic and aromatic diacyl chlorides (sebacoyl, adipoyl, and terephthaloyl). Optimization of reaction conditions (solvent, base nature, and addition mode) has been investigated with protected alcohols. The use of hindered bases was needful to ensure the selectivity of the addition reaction when starting from unprotected aminoalcohol, and selectively led to bis amides products with good yields. 1D and 2D NMR techniques were used to ascertain the structures of the unprecedented monomers. These novel building blocks were successfully polymerized using succinic and terephthalic spacers and studied by NMR and DSC techniques.
1. Introduction
The design of new polymer materials from renewable resources offered an enormous potential to replace the depleting fossil feedstock and can be considered as an environmental alternative.[Citation1] 1,4:3,6-dianhydrohexitols (DAH),[Citation2] three isomers of high importance in this context, were widely used as monomers and building blocks in polymer chemistry.[Citation3–6] More specifically, isosorbide (Is), the most produced monomer on industrial scale, was extensively investigated but suffered from a lack of reactivity on its hydroxyl functions.[Citation7]
Therefore in order to be used in commercial polymers, more reactive 1,4:3,6-dianhydrohexitols derivatives were needed. In this field, several interesting works dealt with their transformation into amines,[Citation8,9] acids,[Citation10] or extended primary alcohols.[Citation11] In this context, the majority of interest was focused on isosorbide as being central core and the modification was mainly considered at both extremities in order to build diesters [Citation12,13] or oligoesters, diglycidyl ethers,[Citation14] diamines,[Citation15–17] diacids[Citation18], and so on.
Another feature regarding isosorbide lies in the difference of reactivity which rendered the obtaining of polymers with stereoregular sequences difficult to reach. One approach to overcome this issue was to obtain novel diols with defined stereochemistry, thanks to the insertion of spacers to establish a new versatile platform. Pioneering works of Thiem and co-workers proposed the synthesis of terephthalic diester triad starting from two equivalents of protected isosorbide.[Citation19] This strategy could pave the way for building copolymers with a great flexibility and regular sequence.[Citation20] Actually it will be possible starting from a diol building block to take benefit from the properties of both two monomers.[Citation21] In this way, the latter could be aliphatic or aromatic and linked by several types of functions such as amides, ester, urethane. To the best of our knowledge, in addition to Thiem works, only one paper reported such strategy with diurethane isosorbide-based diols.[Citation22]
We recently reported the synthesis of poly(ether)ester-amides (PeEA) obtained from aminoalcohols based on isosorbide and isomannide, two isomers of 1,4:3,6-dianhydrohexitols.[Citation23] The first step involved a solvent-free SNAr affording mononitrophenyl–dianhydrohexitol intermediate followed by a reduction reaction. The obtained polymers exhibited interesting properties allying the ester and amide bond characters but the sequences were random affecting their thermal properties. Moreover, we could not vary the spacer nature in the same polymer and control their thermal or mechanical behavior.
In this paper, we envision to build an unprecedented platform of segmented biosourced diols. To ensure this goal, an iterative strategy is initiated in order to undergo selective transformation starting from exo-protected isosorbide monomer to obtain a bis-amide-based platform (BAm_Is). The strategy involves a monoarylation of the latter to obtain the corresponding nitrophenyl-protected alcohol which represents the starting precursor. A selective reduction in the nitro function would give the amine leading to the desired bis-amide. We expect to retain the rigidity in this manner, while significantly increasing the further reactivity of the building block, thanks to the less hindered OH as functional groups. These unprecedented diols are reacted in bulk with either succinic acid or terephthalic chloride in order to obtain a copolymer with tuneable thermal properties.
2. Experimental
2.1 Materials and methods
Isosorbide (98%) was purchased from Alfa Aeser (Karlsruhe, Germany). 1,4,7,10,13,16-Hexaoxacyclooctadecane (18-Crown-6), 1-fluoro-4-nitrobenzene (99%), activated palladium supported on charcoal (10%), sebacoyl chloride (99%), suberoyl chloride (97%), adipoyl chloride (98%), terephthaloyl chloride (≥99%), isophthaloyl chloride (≥99%), pyridine (anhydrous, 99.8%), 2,3,5-collidine (99%), and 2,6-lutidine were purchased from Sigma-Aldrich (St Louis, MO, USA). Hydrazine monohydrate (98%) was purchased from Loba Chemie (Mumbai, India) and KOH (Normapur) was purchased from Prolabo (Paris, France). All solvents and pyridine were distilled over P2O5 and CaH2, respectively, and placed on molecular sieves.
1D and 2D NMR techniques were recorded at 500 MHz (Bruker WP 250). The chemical shifts are given in ppm. Mass spectrometry (MS) was performed on a Autospec Ultima (Waters/Micromass) device equipped with an electrospray ionization (ESI) ion source in positive mode.
2.2. Synthesis of protected alcohol
2.2.1. (3S,3aR,6R,6aR)-3-(benzyloxy)-6-(4-nitrophenoxy)hexahydrofuro[3,2-b]furan (1)
(3R,3aR,6S,6aR)-6-(benzyloxy)hexahydrofuro[3,2-b]furan-3-ol (3.17 mmol, 750 mg), KOH (4.81 mmol, 270 mg) p-fluoronitrobenzene (4.14 mmol, 585 mg) and 18-Crown-6 (10%, 84 mg) were placed in a cylindrical reactor equipped with a mechanical stirrer. The reaction media was warmed gradually to 140 °C and stirring was maintained during 3 h. After reaction completion, the crude product was diluted with chloroform and precipitated in cold methanol, affording the nitro compound 1 after filtration as a yellow solid (911 mg, 81%). 1H NMR (ppm, CDCl3), δ = 8.24–8.19 (d, 2H, H14, J = 9.22 Hz), 7.34 (m, 5H, H9–11), 7.05–7.00 (d, 2H, H13, J = 9.22 Hz), 5.03–4.98 (t, 1H, H4, J = 5.05 Hz), 4.90–4.83 (dd, 1H, H5, J = 9.98 Hz, J′ = 4.93 Hz), 4.64–4.61 (d, 1H, H3, J = 4.93 Hz), 4.59 (s, 2H, H7), 4.16–4.14 (d, 1H, H2, J = 3.16 Hz), 4.03–3.99 (m, 3H, H6 + H1), 3.93–3.86 (dd, 1H, H1, J = 10.11 Hz, J′ = 3.66 Hz); 13C NMR (ppm, CDCl3) δ = 163.04 (C-12), 141.94 (C-15), 137.42 (C-8), 128.52 (C-9), 127.95 (C-10), 127.72 (C-11), 125.88 (C-14), 115.09 (C-13), 86.89 (C-3), 82.98 (C-2), 80.99 (C-4), 78.26 (C-5), 73.30 (C-1), 71.48 (C-7), 71.15 (C-6).
2.2.2. 4-(((3R,3aR,6S,6aR)-6-(benzyloxy)hexahydrofuro[3,2-b]furan-3-yl)oxy)aniline (2)
1 (1.96 mmol, 700 mg) was dissolved in EtOH (31 mL) with Pd/C (10%, 70 mg) in two round-bottom necked flasks. The media was warmed to 50 °C; then a hydrazine solution (39.2 mmol, 1.9 mL in 5 mL of EtOH) was added dropwise for 1 h. When addition was completed, the reaction was refluxed for an additional 2 h. After completion, the warm crude was filtered on an ice bath and the product 2 was recovered by precipitation in EtOH as a pure white crystalline solid (510 mg, 80%). 1H NMR (ppm, CDCl3), δ = 7.36–7.30 (m, 5H, H9–11), 6.85–6.82 (d, 2H, H13, J = 8.67 Hz), 6.64–6.61 (d, 2H, H14, J = 8.86 Hz), 4.84–4.81 (t, 1H, H4, J = 4.62 Hz), 4.65–4.59 (m, 4H, H5 + H3 + H7), 4.14–4.12 (dd, 1H, H2, J = 3.08 Hz, J′ = 2.31 Hz), 4.07–4.02 (m, 3H, H1 + H6), 3.85–3.80 (dd, 1H, H6, J = 9.06 Hz, J′ = 7.13 Hz), 3.46 (s, 2H, NH2); 13C NMR (ppm, CDCl3) δ = 150.99 (C-12), 140.95 (C-15), 137.55 (C-8), 128.45 (C-9), 127.82 (C-11), 127.68 (C-10), 117.27 (C-14), 116.21 (C-13), 86.42 (C-3), 83.62 (C-2), 80.64 (C-4), 79.41 (C-5), 73.38 (C-1), 71.45 (C-7), 70.05 (C-6).
2.3. General procedure for addition reaction
2.3.1. N1,N10-bis(4-(((3R,3aR,6S,6aR)-6-(benzyloxy)hexahydrofuro[3,2-b]furan-3-yl)oxy)phenyl)decanediamide (3a)
A solution of pyridine (1.21 mmol, 98 μL in 3 mL of CH2Cl2) was added dropwise to a solution of 2 (1.22 mmol, 400 mg) in 10 mL of CH2Cl2. When addition was completed, the media was cooled at 0 °C then a solution of sebacoyl chloride (0.6 mmol, 128 μL in 3 mL of CH2Cl2) was added dropwise for 1 h. During the addition, a precipitate was formed immediately. After reaction completion, the crude was filtered and washed with distilled water affording the adduct 3a as a brown solid (433 mg, 88%). 1H NMR (ppm, DMSO-d6), δ = 9.70 (s, 2H, Ha), 7.47–7.45 (d, 4H, H14, J = 8.83 Hz), 7.37–7.28 (m, 10H, H9–11), 6.93–6.91 (d, 4H, H13, J = 9.14 Hz), 4.84–4.82 (t, 2H, H4, J = 4.73 Hz), 4.82–4.79 (q, 2H, H5, J = 5.36 Hz), 4.57–4.51 (m, 6H, H3 + H7), 4.05 (d, 2H, H2, J = 3.15 Hz), 3.97–3.94 (dd, 2H, H6′, J = 9.14 Hz, J′ = 5.99 Hz), 3.88–3.86 (d, 2H, H1′, J = 10.09 Hz), 3.81–3.78 (dd, 2H, H1, J = 10.09 Hz, J′ = 3.47 Hz), 3.70–3.67 (dd, 2H, H6, J = 9.46 Hz, J′ = 5.67 Hz), 2.26–2.23 (t, 4H, Hb, J = 7.57 Hz), 1.58–1.56 (t, 4H, Hc, J = 5.67 Hz), 1.29 (s, 8H, He + Hd); 13C NMR (ppm, DMSO-d6) δ = 170.73 (C-f), 153.47 (C-12), 138.00 (C-8), 132.78 (C-15), 128.25 (C-9), 127.64 (C-10), 127.52 (C-11), 120.43 (C-14), 115.09 (C-13), 85.79 (C-3), 83.00 (C-2), 80.04 (C-4), 77.25 (C-5), 72.49 (C-1), 70.26 (C-7), 69.96 (C-6), 36.25 (C-b), 28.64 (C-d + C-e), 25.15 (C-c); ESI-MS: (M + Na)+: C48H56N2NaO10 843.4 g mol−1.
2.3.2. N1,N8-bis(4-(((3R,3aR,6S,6aR)-6-(benzyloxy)hexahydrofuro[3,2-b]furan-3-yl)oxy)phenyl)octanediamide (3b)
The general procedure was applied to a solution of pyridine (1.21 mmol, 98 μL), 2 (1.26 mmol, 413 mg), and a solution of suberoyl chloride (0.63 mmol, 126 μL) to obtain 3b as a brown solid (225 mg, 45%). 1H NMR (ppm, DMSO-d6), δ = 9.73 (s, 2H, Ha), 7.47–7.45 (d, 4H, H14, J = 8.83 Hz), 7.36–7.27 (m, 10H, H9–11), 6.92–6.91 (d, 4H, H13, J = 8.83 Hz), 4.84–4.82 (t, 2H, H4, J = 4.73 Hz), 4.82–4.78 (dd, 2H, H5, J = 11.03 Hz, J′ = 5.36 Hz), 4.56–4.51 (m, 6H, H3 + H7), 4.05–4.04 (d, 2H, H2, J = 2.84 Hz), 3.97–3.94 (dd, 2H, H6′, J = 9.14 Hz, J′ = 5.67 Hz), 3.88–3.86 (d, 2H, H1′, J = 10.09 Hz), 3.80–3.77 (dd, 2H, H1, J = 9.77 Hz, J′ = 3.78 Hz), 3.70–3.67 (dd, 2H, H6, J = 9.14 Hz, J′ = 5.67 Hz), 2.27–2.24 (t, 4H, Hb, J = 7.25 Hz), 1.59–1.57 (m, 4H, Hc), 1.31 (s, 4H, Hd); 13C NMR (ppm, DMSO-d6) δ = 170.78 (C-f), 153.52 (C-12), 138.04 (C-8), 132.81 (C-15), 128.31 (C-9), 127.70 (C-10), 127.59 (C-11), 120.50 (C-14), 115.06 (C-13), 85.83 (C-3), 83.02 (C-2), 80.09 (C-4), 77.28 (C-5), 72.53 (C-1), 70.29 (C-7), 70.02 (C-6), 36.27 (C-b), 28.53 (C-d), 25.14 (C-c); ESI-MS: (M + Na)+, C46H52N2NaO10 : 815.4 g mol−1.
2.3.3. N1,N6-bis(4-(((3R,3aR,6S,6aR)-6-(benzyloxy)hexahydrofuro[3,2-b]furan-3-yl)oxy)phenyl)adipamide (3c)
The general procedure was applied to a solution of pyridine (1.00 mmol, 81 μL), 2 (1.03 mmol, 337 mg), and a solution of adipoyl chloride (0.50 mmol, 73 μL) to obtain 3c as a brown solid (210 mg, 55%). 1H NMR (ppm, DMSO-d6), δ = 9.72 (s, 2H, Ha), 7.47–7.45 (d, 4H, H14, J = 9.14 Hz), 7.36–7.27 (m, 10H, H9–11), 6.92–6.90 (d, 4H, H13, J = 8.83 Hz), 4.84–4.82 (t, 2H, H4, J = 4.73 Hz), 4.81–4.78 (q, 2H, H5, J = 5.36 Hz), 4.56–4.50 (m, 6H, H3 + H7), 4.04 (d, 2H, H2, J = 3.47 Hz), 3.96–3.93 (dd, 2H, H6′, J = 9.46 Hz, J′ = 5.67 Hz), 3.88–3.86 (d, 2H, H1′, J = 10.09 Hz), 3.80–3.77 (dd, 2H, H1, J = 10.09 Hz, J′ = 3.47 Hz), 3.69–3.66 (dd, 2H, H6, J = 9.14 Hz, J′ = 5.67 Hz), 2.28 (s, 4H, Hb), 1.61 (s, 4H, Hc); 13C NMR (ppm, DMSO-d6) δ = 170.61 (C-f), 153.53 (C-12), 138.03 (C-8), 132.76 (C-15), 128.30 (C-9), 127.69 (C-10), 127.58 (C-11), 120.49 (C-14), 115.06 (C-13), 85.81 (C-3), 83.02 (C-2), 80.08 (C-4), 77.27 (C-5), 72.52 (C-1), 70.28 (C-7), 70.00 (C-6), 36.18 (C-b), 25.00 (C-c); ESI-MS: (M + Na)+, C44H48N2NaO10 787.4 g mol−1.
2.3.4. N1,N4-bis(4-(((3R,3aR,6S,6aR)-6-(benzyloxy)hexahydrofuro[3,2-b]furan-3-yl)oxy)phenyl)terephthalamide (3d)
The general procedure was applied to a solution of pyridine (1.22 mmol, 99 μL), 2 (1.26 mmol, 414 mg), and a solution of terephthaloyl chloride (0.60 mmol, 122 mg) to obtain 3d as a brown solid (376 mg, 80%). 1H NMR (ppm, DMSO-d6), δ = 10.26 (s, 2H, Ha), 8.07 (s, 4H, Hb + Hc), 7.68–7.66 (d, 4H, H14, J = 9.14 Hz), 7.37–7.28 (m, 10H, H9–11), 7.02–7.00 (d, 4H, H13, J = 8.83 Hz), 4.89–4.85 (m, 4H, H4 + H5), 4.58–4.52 (m, 6H, H3 + H7),4.07–4.06 (d, 2H, H2, J = 3.47 Hz), 4.04–3.97 (dd, 2H, H6′, J = 8.83 Hz, J′ = 5.67 Hz), 3.90–3.88 (d, 2H, H1′, J = 10.09 Hz), 3.83–3.80 (dd, 2H, H1, J = 10.09 Hz, J′ = 3.78 Hz), 3.74–3.71 (dd, 2H, H6, J = 8.83 Hz, J′ = 5.36 Hz); 13C NMR (ppm, DMSO-d6) δ = 164.37 (C-f), 154.24 (C-12), 138.01 (C-8), 137.36 (C-b), 132.24 (C-15), 128.27 (C-9), 127.67 (C-10), 127.58 (C-11), 127.54 (C-c), 121.96 (C-14), 115.01 (C-13), 85.84 (C-3), 83.00 (C-2), 80.11 (C-4), 77.26 (C-5), 72.52 (C-1), 70.28 (C-7), 70.06 (C-6); ESI-MS: (M + Na)+, C46H44N2NaO10 807.3 g mol−1.
2.3.5. N1,N3-bis(4-(((3R,3aR,6S,6aR)-6-(benzyloxy)hexahydrofuro[3,2-b]furan-3-yl)oxy)phenyl)isophthalamide (3e)
The general procedure was applied to a solution of pyridine (1.05 mmol, 85 μL), 2 (1.05 mmol, 345 mg), and a solution of terephthaloyl chloride (0.50 mmol, 101.50 mg) to obtain 3e as a brown solid (282 mg, 72%). 1H NMR (ppm, DMSO-d6), δ = 10.32 (s, 2H, Ha), 8.51 (s, 1H, Hb), 8.14–8.12 (d, 2H, Hc, J = 7.57 Hz), 7.70–7.68 (d, 4H, H14, J = 8.83 Hz), 7.69–7.67 (d, 1H, Hd, J = 7.25 Hz), 7.39–7.30 (m, 10H, H9–11), 7.04–7.02 (d, 4H, H13, J = 9.14 Hz), 4.91–4.86 (m, 4H, H4 + H5), 4.61–4.54 (m, 6H, H3 + H7),4.08–4.07 (d, 2H, H2, J = 3.47 Hz), 4.01–3.98 (dd, 2H, H6′, J = 9.14 Hz, J′ = 5.67 Hz), 3.92–3.90 (d, 2H, H1′, J = 10.09 Hz), 3.84–3.81 (dd, 2H, H1, J = 10.09 Hz, J′ = 3.47 Hz), 3.76–3.73 (dd, 2H, H6, J = 9.14 Hz, J′ = 5.36 Hz); 13C NMR (ppm, DMSO-d6) δ = 164.69 (C-f), 154.20 (C-12), 138.04 (C-8), 135.26 (C-e), 132.35 (C-15), 130.41 (C-b), 128.58 (C-c), 128.30 (C-9), 127.70 (C-10), 127.58 (C-11), 126.82 (C-d), 121.88 (C-14), 115.04 (C-13), 85.86 (C-3), 83.02 (C-2), 80.12 (C-4), 77.27 (C-5), 72.54 (C-1), 70.29 (C-7), 70.08 (C-6); ESI-MS: (M + Na)+, C46H44N2NaO10 : 807.3 g mol−1.
2.4. (3S,3aR,6R,6aR)-6-(4-aminophenoxy)hexahydrofuro[3,2-b]furan-3-ol (4)
Monomer synthesis and characterization are provided in details in this article.[Citation23]
2.5. General procedure for condensation of 4 with diacyl chloride
2.5.1. N1,N10-bis(4-(((3R,3aR,6S,6aR)-6-hydroxyhexahydrofuro[3,2-b]furan-3-yl)oxy)phenyl)decanediamide (5a)
The aminoalcohol 4 (0.84 mmol, 200 mg) was dissolved in 5 mL of CH2Cl2. After dissolution, the media was cooled at 0 °C and a solution of 2,3,5-collidine (1.68 mmol, 219 μL in 1 mL of CH2Cl2) (or 1.68 mmol of 2,6-Lutidine) and a solution of sebacoyl chloride (0.42 mmol, 87 μL in 1 mL of CH2Cl2) were simultaneously added dropwise. After addition completion, a precipitate was observed and the reaction was maintained over the night. The crude was filtered and washed with distilled water to afford the adduct 5a as a white solid (141 mg, 53%) (5d, brown solid, 183 mg, 69%). 1H NMR (ppm, DMSO-d6), δ = 9.72 (s, 2H, Ha), 7.48–7.46 (d, 4H, H14, J = 8.83 Hz), 6.93–6.92 (d, 4H, H13, J = 9.14 Hz), 5.21 (s, 2H, OH), 4.82–4.78 (m, 4H, H4 + H5), 4.35–4.34 (d, 2H, H3, J = 3.78 Hz), 4.11–4.10 (d, 2H, H2, J = 3.47 Hz), 3.97–3.94 (dd, 2H, H6′, J = 8.83 Hz, J′ = 5.99 Hz), 3.79–3.76 (dd, 2H, H1, J = 9.46 Hz, J′ = 3.47 Hz), 3.68–3.63 (m, 4H, H1′ + H6), 2.28–2.25 (t, 4H, Hb, J = 7.57 Hz), 1.60–1.57 (t, 4H, Hc, J = 6.31 Hz), 1.30 (s, 8H, Hd + He); 13C NMR (ppm, DMSO-d6) δ = 170.88 (C-f), 153.61 (C-12), 132.77 (C-15), 120.57 (C-14), 115.12 (C-13), 88.49 (C-3), 79.80 (C-4), 77.48 (C-5), 75.32 (C-2), 75.24 (C-1), 69.77 (C-6), 36.32 (C-b), 28.74 (C-d), 28.70 (C-e), 25.23 (C-c); ESI-MS: (M + Na)+, C34H44N2NaO10 : 663.3 g mol−1.
2.5.2. N1,N6-bis(4-(((3R,3aR,6S,6aR)-6-hydroxyhexahydrofuro[3,2-b]furan-3-yl)oxy)phenyl)adipamide (5b)
The general procedure was applied to 4 (0.84 mmol, 200 mg), 2,3,5-collidine (1.68 mmol, 219 μL in 1 mL of CH2Cl2) (or 1.68 mmol of 2,6-Lutidine), and adipoyl chloride (0.42 mmol, 67 μL in 1 mL of CH2Cl2) to afford 5b as a brown solid (187 mg, 76%) (5e, brown solid, 125 mg, 51%). 1H NMR (ppm, DMSO-d6), δ = 9.78 (s, 2H, Ha), 7.46–7.43 (d, 4H, H14, J = 8.88 Hz), 6.92–6.89 (d, 4H, H13, J = 9.06 Hz), 5.27 (s, 2H, OH), 4.84–4.76 (m, 4H, H4 + H5), 4.32–4.31 (d, 2H, H3, J = 2.83 Hz), 4.09–4.08 (d, 2H, H2, J = 2.27 Hz), 3.95–3.90 (dd, 2H, H6′, J = 8.69 Hz, J′ = 5.67 Hz), 3.76–3.72 (dd, 2H, H1, J = 9.44 Hz, J′ = 3.40 Hz), 3.66–3.61 (m, 4H, H1′ + H6), 2.28 (s, 4H, Hb), 1.60 (s, 4H, Hc); 13C NMR (ppm, DMSO-d6) δ = 170.98 (C-f), 153.80 (C-12), 132.79 (C-15), 120.79 (C-14), 115.25 (C-13), 88.58 (C-3), 79.96 (C-4), 77.55 (C-5), 75.43 (C-2), 75.34 (C-1), 69.95 (C-6), 36.33 (C-b), 25.18 (C-c); ESI-MS: (M + Na)+, C30H36N2NaO10 : 607.3 g mol−1.
2.5.3. N1,N4-bis(4-(((3R,3aR,6S,6aR)-6-hydroxyhexahydrofuro[3,2-b]furan-3-yl)oxy)phenyl)terephthalamide (5c)
The general procedure was applied to 4 (0.84 mmol, 200 mg), 2,3,5-collidine (1.68 mmol, 219 μL in 1 mL of CH2Cl2) (or 1.68 mmol of 2,6-Lutidine), and terephthaloyl chloride (0.42 mmol, 85.26 mg in 1 mL of CH2Cl2) to afford 5c as a white solid (212 mg, 84%) (5f, brown solid, 193 mg, 77%). 1H NMR (ppm, DMSO-d6), δ = 10.31 (s, 2H, Ha), 7.48–7.46 (d, 4H, H, J = Hz), 6.93–6.92 (d, 4H, H, J = Hz), 5.21 (s, 2H, H), 4.824.78 (m, 4H, H), 4.35–4.34 (d, 2H, H, J = Hz), 4.11–4.10 (d, 2H, H, J = Hz), 3.97–3.94 (dd, 2H, H, J = Hz, J′ = Hz), 3.79–3.76 (dd, 2H, H, J = Hz, J′ = Hz), 3.68–3.63 (m, 4H, H), 2.28–2.25 (t, 4H, H, J = Hz), 1.60–1.57 (t, 4H, H, J = Hz), 1.30 (s, 8H, H); 13C NMR (ppm, DMSO-d6) δ = 170.88 (C-f), 153.61 (C-12), 132.77 (C-15), 120.57 (C-14), 115.12 (C-13), 88.49 (C-3), 79.80 (C-2), 77.48 (C-4), 75.32 (C-5), 75.24 (C-1), 69.77 (C-6), 36.32 (C-b), 28.74 (C-d), 28.70 (C-e), 25.23 (C-c); ESI-MS: (M + Na)+: 627.2 g mol−1.
2.6. General procedure for polymerization
2.6.1. Synthesis of Pe_EA 8
5a (0.51 mmol, 330 mg) and succinic acid 6 (0.51 mmol, 61 mg) were charged into a round-bottom flask with a mechanical stirring. The apparatus was flushed with nitrogen flow to remove oxygen and then heated at 175 °C and a homogeneous melt was obtained. Sn(oct)2 (1% wt) was added under nitrogen atmosphere and a pre-polymerization was carried on. The polymerization temperature was then increased to 200 °C and the polymerization was allowed to undergo for 3 h. After cooling down to room temperature, the crude was dissolved in DMF and precipitated into cold MeOH giving a brown solid. 1H NMR (ppm, DMSO-d6), δ = 12.12 (s, Hi), 9.75 (s, 2H, Ha), 7.49–7.47 (d, 4H, H14), 6.92–6.90 (d, 4H, H13), 5.21 (s, Hj), 5.07 (s, 2H, H2), 4.86–4.79 (m, 4H, H4 + H5), 4.46–4.45 (d, 2H, H3), 4.33 (s, H3*), 4.09 (s, H2*), 3.94–3.74 (m, 8H, H6 + H1), 3.66–3.61 (m, H1*), 2.60–2.57 (d, 4H, Hh), 2.42 (s, Hh*), 2.25 (m, 4H, Hb), 1.56 (m, 4H, Hc), 1.28 (s, 8H, Hd + He); 13C NMR (ppm, DMSO-d6) δ = 177.84 (C-g*), 173.72 (C-g), 171.35 (C-f*), 170.86 (C-f), 153.46 (C-12), 132.92 (C-15), 120.50 (C-14), 115.06 (C-13), 88.50 (C-3*), 85.69 (C-3), 80.37 (C-4), 79.80 (C-4*), 78.09 (C-2), 77.46 (C-5*), 77.07 (C-5), 75.33 (C-2*), 75.25 (C-1*), 72.53 (C-1), 69.83 (C-6); 69.77 (C-6*), 37.73 (C-h), 36.32 (C-b), 33.40 (C-b*), 28.79 (C-d), 28.75 (C-e), 28.42 (C-d*), 28.03 (C-e*), 25.76 (C-c*), 25.26 (C-c).
2.6.2. Synthesis of Pe_EA 9
The general procedure for polymerization was applied to 5a (0.75 mmol, 483 mg), terephthaloyl chloride 7 (0.83 mmol, 168.40 mg), and Sn(oct)2 (1% wt) to obtain 9 as a black solid.1H NMR (ppm, DMSO-d6), δ = 10.33–10.28 (m, Hi), 9.71 (s, 2H, Ha), 8.08 (s, 4H, Hk + Hl), 7.69–7.66 (m, H*), 7.47–7.46 (d, 4H, H14), 7.03–7.00 (m, H*), 6.92–6.90 (d, 4H, H13), 5.37–5.35 (d, Hj), 5.08–5.06 (d, 2H, H2), 4.91–4.79 (m, 4H, H4 + H5), 4.70 (s, H2*), 4.46–4.45 (d, 2H, H3), 4.33 (s, H3*), 4.04–3.71 (m, 8H, H6 + H1), 2.28–2.23 (m, 4H, Hb), 1.56–150 (m, 4H, Hc), 1.28–1.23 (m, 8H, Hd + He); 13C NMR (ppm, DMSO-d6) δ = 172.23 (C-f*), 170.72 (C-f), 166.61 (C-g*), 162.26 (C-g), 153.39 (C-12), 134.42 (C-15), 129.39 (Ck + Cl), 127.55 (C*), 121.94 (C*), 120.44 (C-14), 115.04 (C-13), 85.73 (C-3), 80.24 (C-4), 77.68 (C-2), 77.11 (C-5), 72.57 (C-1), 70.30 (C-6), 36.25 (C-b), 33.32 (C-b*), 30.73–28.24 (C-d + d* + e + e*), 25.13 (C-c), 24.24 (C-c*).
3. Results and discussion
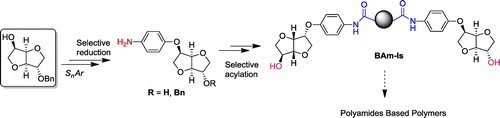
For a selective monoarylation of the diol, a prior protection of one hydroxyl function with classical monobenzylation was needed (Scheme ).[Citation24] Monobenzylated isosorbide and 4-fluoronitrobenzene were reacted in melt in presence of KOH as an alkaline base and 18-Crown-6 as a phase-transfer agent. After precipitation in cold methanol, the nitro compound 1 was isolated as a yellow crystalline powder with high yield (85%). This compound was a key intermediate around which articulated our strategy (Scheme ).
To investigate the feasibility of this strategy a template molecule was needed. Using a difunctional monomer (i.e. aminoalcohol) would not be suitable for a bis-acylation only in one side under classical conditions. Indeed, we recently used the corresponding aminoalcohol as starting monomers for the synthesis of poly(ether)ester-amides by reaction with several diacyl chlorides in presence of pyridine as a base. Several parameters were studied such as base addition effect and in all cases we observed the occurrence of both amidification and esterification. The challenge encountered at this stage was to selectively transform only the nitro functionality into amine for its further use and ensure further deprotection of the alcohol. The reduction step was carefully investigated. Surprisingly working under atmospheric pressure of H2 led to a mixture of products with a majority of amine intermediate. This result was encouraging as it paved the way for a selective reduction. After several optimization tests, the use of hydrazine [Citation25] as source of hydride coupled with catalytic Pd/C in Ethanol successfully afforded after precipitation the amino compound 2 as a white crystalline solid in high isolated yield (80%) while keeping exo alcohol protected. To investigate the chemical structure of the obtained 1,4:3,6-dianhydrohexitols derivatives 1 and 2 and to prove the efficiency of the transformation of nitro into amine, liquid-state NMR was applied. It is noteworthy to mention that the reduction reaction induced a chemical shift for the upfield zone due to the electron-donating effect of NH2 group. Indeed, the group of signals observed in 1H spectra at δH 3.88–5.03 ppm for 1 and 3.77–4.84 ppm for 2 (Figure S1 in the Supporting Information) provided a proof of the efficient chimiospecifity of hydrazine method. While signal of H5 corresponding to nitro compound appeared at 4.86 ppm, the signal of H5 corresponding to amine derivative appeared at 4.63 ppm, showing the modification in electron density around the dianhydrohexitol core. We also observed an inversion of signals of aromatic protons corresponding to H13/H14 between the nitrophenyl alcohol 1 and the corresponding amine 2 at 7.02/8.22 ppm and 6.83/6.63 ppm respectively.
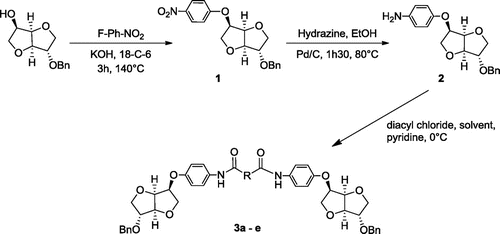
Starting from this compound, we investigated the introduction of spacers by reaction with diacyl chlorides. To ensure the bisacylation to take place the diacyl chloride was added dropwise at 0 °C on a solution of the amino compound 2 in the presence of pyridine as HCl acceptor. After addition completion, the product was isolated by precipitation. Systematic solvent study was undertaken and the best results were depicted in the Table .
Table 1. Exemplification of Bam_Is 3a–e.
Several alkyl chain long have been tested as well as aromatic moieties affording diamides with fair to high yields depending on the solubility of the obtained substrate. As an example for the isophthaloyl compound a precipitation in water was needed to isolate the desired product. This diol precursor, after further deprotection, could be used for the elaboration of versatile copolymers using different bifunctionnal monomers.
Thanks to these preliminary results we did successfully characterize the obtained products by mass spectroscopy and NMR techniques. Starting from the analytical NMR data provided by Hopton and Thomas [Citation26] and based on the analysis of 1H spectrum (Figure ) supported by the single quantum 1H/13C correlations in the HSQC (Figure S2 in the Supporting Information) we efficiently ascertained all the signals of the obtained monomers. As an example, the compound 3a was carefully investigated and revealed interesting findings. Indeed characteristic signals of Is core were complex and the correlation with carbons allowed the right attribution as well as for the aliphatic part of the spacer. In fact, the group of signals at δH 4.84–3.67 ppm provided information about the protons of Is core. We observed the presence of signals corresponding to H3 as a doublet overlapped by H7 at δH 4.57–4.51 ppm confirming the exo position of H3 relative to H4. We observed also the signals of H6′, H1′, H1, and H6 at δH 3.97–3.67 ppm. For the aromatic protons, the multiplet corresponding to the benzyl group appeared at δH 7.37–7.28 ppm and the signals of H14 and H13 appeared respectively at δH 7.46 and 6.92 ppm as an asymmetric doublets integrated each one for two protons showing the AB spin system. It is worthy to mention the presence of signals corresponding to protons of the amide function newly formed at δH 9.70 ppm. Signals of protons corresponding to the sebacoyl chloride appeared at δH 2.26–1.29 ppm. Based on 1H NMR shifts previously detailed, the analysis of 2D correlations provided the tentative attribution of both aromatic and aliphatic carbons (Figure S2 in the Supporting Information). We observed correlations signals of the aliphatic protons corresponding to sebacoyl chloride Hd + e/Cd + e, Hc/Cc and Hb/Cb, respectively, at 1.31/29.16, 1.59/25.10 and 2.26/36.32 ppm. Furthermore, the correlation signals H7/C7, H6/C6, H1/C1, H2/C2, H3/C3, H5/C5 and H4/C4 of Is block at 4.55/69.58, 3.69/69.98, 3.80/72.38, 4.06/82.80, 4.57/85.61, 4.80/77.30, and 4.83/80.00 ppm, respectively, confirmed the attribution of carbons of Is core. The examination of the correlations signals H13/C13 and H14/C14 at 6.92/114.86 and 7.47/120.06 ppm proved the formation of the amide function with less pronounced shifting than the previously detected with nitro 1 and amino 2 compounds.
Figure 1. 1H NMR spectrum of the protected alcohol 3a.
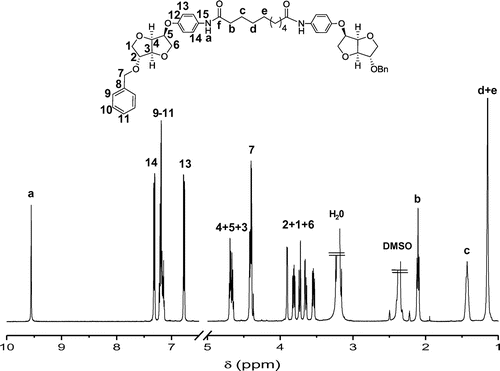
Figure 2. 500 MHz correlation spectrum (COSY) of the diol 5a.
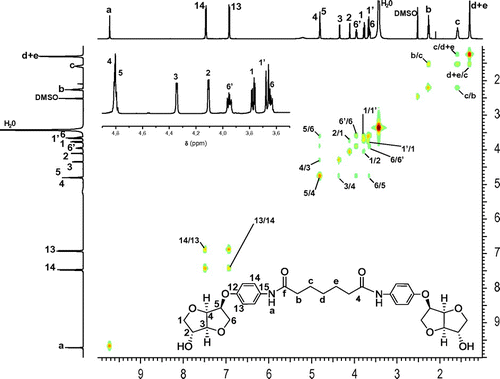
Figure 3. 500 MHz heteronuclear multiple-bond correlation spectrum (gHMBC) of the diol 5a.
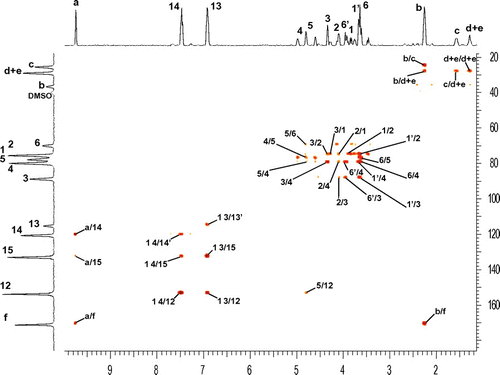
Figure 4. 1H spectra of 5a (B), Pe_EAs 8 (C) and 9 (D).
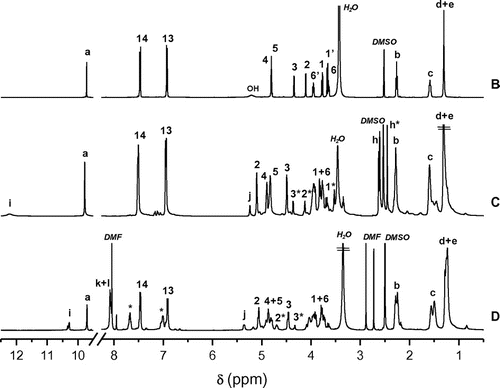
Figure 5. 13C spectra of 5a (E), Pe_EA 8 (F) and Pe_EA 9 (G).
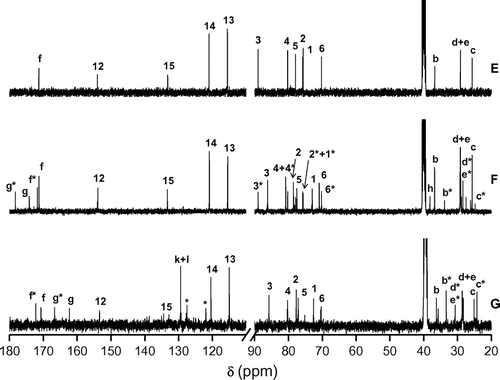
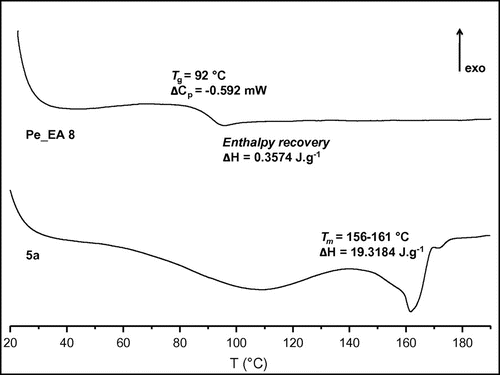
Figure 6. Thermal properties of Pe_EA 8 and diol 5a measured by DSC.
Scheme 1. Synthetic route of novel monomers issued from Isosorbide.
Scheme 2. Synthesis of protected alcohols 3a–e.
Having characterized these premonomers and checked the feasibility of the double addition, we decided to reconsider our strategy to start directly from the corresponding aminoalcohol in order to obtain the Bam_Is diol in fewer steps. The corresponding aminoalcohol, was obtained by one-pot reduction–debenzylation of 1 using 5 bar of H2 catalyzed by Pd/C in ethyl acetate. We previously showed that the base could play a double role of HCl acceptor and catalyst for the esterification by addition on the carbonyl function. The novelty consisted then in decreasing the base’s nucleophilic character to the benefit of the basic one which could selectively advantages only amidification due to the sterical hindrance. To assess this assumption collidine and lutidine were tested (Scheme ). The addition of the latter simultaneously with the diacyl chloride on the aminoalcohol in solution gave rise to the desired Bam_Is by precipitation. Solubility of the obtained Bam_Is did not change comparing to the corresponding protected alcohols due to the intermolecular hydrogen bonds established between the amide groups. Aliphatic spacers as well as aromatic ones were tested and good yields were successfully obtained. Reaction conditions and yields are collected in the Table .
Scheme 3. The strategy for the synthesis of novel diols 5a-c.
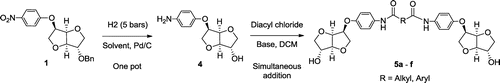
Table 2. Optimization of reaction parameters and yields of diols 5a-c.
These diols were characterized in details by mass spectroscopy and NMR techniques. The examination of 1H and 13C NMR data, supported by the analysis of the long-range 1H/13C correlations in the gHMBC and HSQC spectra, and the cross peak in the COSY technique (Figure ), allowed the rigorous attribution and ascertainment of each proton and carbon.
The study conducted previously on 3a was useful to ascertain all the signals of diol 5a (Figure S4 in the supporting information). Based on the attribution of 3a previously detailed, we observed the absence of signals corresponding to protons of benzyl group at δH 7.37–7.28 and 4.55 ppm. Signal of Ha corresponding to the proton of amide function was detected at δH 9.72 ppm with a slight shift comparing to 3a detected at δH 9.70 ppm. For the signals of protons of Is core, H4 was found to couple with H5 and H3 giving a triplet at δH 4.81 ppm. The cross peaks H4/H5 and H4/H3 observed in the COSY spectrum at δH 4.80 and 4.35 ppm confirmed that the protons H3, H4 and H5 adopted an exo configuration (Figure ). Also signal corresponding to H5 was detected at δH 4.79 ppm as a doublet of doublets, showing the interaction between H5 and respectively H4 and H6 with different constant of coupling 3J5–4 and 3J5–6. This observation proved the fact that one of protons H6 occupied the exo configuration. Examination of the cross-peaks H3/H4 and H2/H1 of COSY spectrum at δH 4.35 and 4.11 ppm respectively, was a proof of the interesting shifts of H3 to upfield zone instead of downfield one for H2 comparing to 3a. This may be influenced by the absence of benzyl group. A slight shift of signals corresponding to the aromatic protons of the aminoalcohol as well as the aliphatic ones issued from sebacoyl chloride was detected. As an example, signals of H14 and Hd + e of 3a were observed at δH 7.46 and 1.29 ppm while for 5a they were observed at δH 7.47 and 1.30 ppm, respectively. This unremarkable shift may be explained by the influence of benzyl group on the electronic density that decreased starting from Is core to spacer. It is noteworthy to mention that the absence of a cross peak between H2 and H3 due to their stereochemistry confirmed that H2 occupied the endo configuration. Besides, a remarkable shift of chemical displacement of H1′ was observed from δH 3.88 ppm for 3a to δH 3.68 ppm for 5a (Figure S4 in the supporting information). This observation can be confirmed due to the cross peak H1/H1′ observed at δH 3.77/3.68 ppm. Moreover, a cross peak H6′/H6 was detected at δH 3.96/3.63 ppm.
The long-range 1H/13C correlations observed in the g-HMBC spectrum (Figure and Figure S5 in the Supporting Information) supported by the single 1H/13C correlations shown in the HSQC spectrum (Figure S3 in the Supporting Information) gave us more detailed information about the configuration of the bicyclic frame of isosorbide-based aminoalcohol and confirmed the above mentioned attribution.
In fact, the examination of the gHMBC correlation signals H4/C5, H5/C6, H4/C3 and H5/C4 at 4.82/77.48, 4.80/69.77, 4.82/88.49, and 4.80/79.80 ppm (Figure S5 in the supporting information), respectively, pressed by the HSQC correlations H4/C4 and H5/C5, at 4.82/79.80 and 4.80/77.48 ppm, respectively, confirmed the attribution of signals corresponding to carbons C4 and C5.
Besides, the observation of the gHMBC correlation signals H3/C2, H2/C1, H3/C4, and H2/C3 at 4.35/75.32, 4.11/75.24, 4.35/79.80, and 4.11/88.49 ppm, respectively, supported by the HSQC correlations H3/C3 and H2/C2 at 4.35/88.49 and 4.11/75.32 ppm proved that C2 remarkably shifted to upfield zone while C3 remained the most deshielded carbon of the Is core. Moreover, the consideration of the correlation signals H6′/C4, H1′/C3, H6/C5 and H1/C2 at 3.97/79.80, 3.68/88.49, 3.65/77.48 and 3.79/75.32 ppm, respectively, stressed by the correlations H6′/C6, H1′/C1, H6/C6 and H1/C1 at 3.97/69.77, 3.68/75.24, 3.65/69.77, and 3.79/75.24 ppm each, confirmed that C6 was considered as the most shielded carbon of Is core. Furthermore, the right assignment of aromatic protons and therefore aromatic carbons was possible only through the presence of gHMBC correlation signals (Figure ) Ha/C14 and Ha/C15 found at 9.72/120.57 and 9.72/132.77 ppm, respectively. The absence of correlation of Ha with C12 and C13 was considered as a proof of the assignments of aromatic protons and carbons. For the aliphatic part of the diol 5a, we observed the correlation signals Ha/Cf, Hb/Cc, Hb/Cd + e, Hb/Cf, Hc/Cd + e and Hd + e/Cd + e, respectively, at 9.72/170.88, 2.28/25.23, 2.28/28.74, 2.28/170.88, 1.60/28.74, and 1.30/28.74 ppm showing once again the right attribution detailed above.
Having these unprecedented diols in hand, we investigated their utility as a building block in polymer materials. In this context, we carried out the polymerization of 5a, with either succinic acid or terephthalic chloride, in bulk and in the presence of Tin (II) 2-ethylhexanoate as a catalyst Sn(oct)2 (1% wt) (Scheme ).
Scheme 4. Synthetic route of Pe_EAs 8 and 9 from diol 5a.
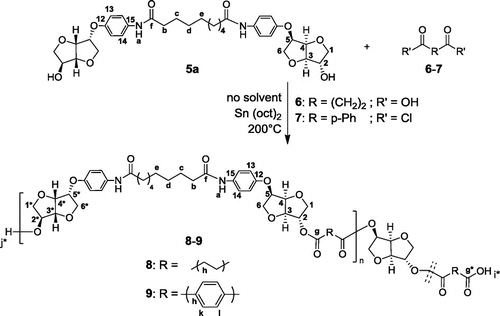
The reaction media was heated at 175 °C to ensure the melting of 5a and then increased to 200 °C to carry on the polycondensation. A precipitation of the crude previously dissolved in DMF, was conducted in cold MeOH leading to solid polymers. To prove the efficiency of our polymerization, 1H and 13C NMR techniques were realized. 1H spectra of Pe_EAs 8 and 9 overlaid with starting monomer 5a were shown in Figure . We observed the presence of signals with high intensity and others with low ones corresponding respectively to the protons from the main chain of the polymer and the end chain moiety. Starting with Pe_EA 8, it is worth mentioning that only signals of isosorbide and succinic acid cores were subjected to main changes in contrast with the aromatic part of the diol. Signals corresponding to H3 and H2 were greatly affected by the esterification when compared with 5a and appeared respectively at 4.46 and 5.07 ppm in the repetitive unit whereas those of the end chain appeared at 4.33 and 4.09 ppm, respectively. Protons Hh of the aliphatic chain of succinic acid appeared at 2.58 ppm for the polymer main chain and 2.42 ppm for the end chain moiety. The chemical shift of signals corresponding to the sebacic spacer H(b-e) remained unchanged in the polymer. Regarding Pe_EA 9, we observed the appearance of signals corresponding to the terephthalic moiety (Hk + Hl) at 8.08 ppm and the displacement of Hi at 10.33 ppm under the influence of the aromatic ring.
Looking carefully to the 13C NMR spectra superposition of monomer 5a and polymer 8 (Figure ), we noticed, besides the signals corresponding to Cf and Cf* of amide functions, the appearance of newly formed carbonyl signals either relative to ester group Cg or carboxylic acid Cg* of the polymer end chain. For Is core, we observed that polymerization gave rise to novel signals corresponding to the main chain polymer. As an example, signals corresponding to C3 and C4 were detected respectively at 85.69 and 80.37 ppm. C1 and C2 were greatly influenced by the change in chemical neighborhood. In fact, they appeared at 72.53 and 78.09 ppm, respectively. This shift was different from one carbon to another depending on its stereochemistry and its location in the polymer chain. The above-mentioned observations about Is core were observed with polymer 9 in addition to novel aromatic carbons of the terephthalic core, where new signals corresponding to Ch, Ck and Cl were obtained at 129.39, 127.55, and 121.94 ppm, respectively. For carbonyl groups, the presence of aromatic ring caused a deshielded effect. For example, while signal corresponding to Cg of 8 was observed at 173.72 ppm, the one related to 9 was detected at 162.26 ppm.
Thermal properties of the polymer Pe_EA 8 and our diol 5a were investigated by DSC analysis (Figure ). Based on our previous works, the use of succinic acid as a spacer aimed to minimize the polyester soft block and to obtain a mainly rigid copolymer. In this context, while 5a had a melting point Tm = 156–161 °C, we observed with Pe_EA 8 a glass transition temperature Tg = 92 °C with an enthalpy recovery ΔH = 19.3184 J g−1 probably due to a physical aging, currently known in sugar chemistry.[Citation27] Comparing with previously obtained semi-crystalline Polyester-amides PeEA[Citation23] fully based on sebacoyl spacer (Tg 72–75 °C), the thermal performance was enhanced since amorphous polymers could be obtained which represent an appealing property for processing. This strategy allows for building copolymers with a great flexibility due to a wide structural flexibility offered by the spacer nature as well as the diacid type used in the polymerization to tune the Tg range.
4. Conclusion
In conclusion, new biosourced diols with amide and alcohol functionalities were synthesized starting from aminoalcohol based on isosorbide. Starting from one-pot catalytic hydrogenation of the nitro function and the protected alcohol followed by condensation with several aliphatic and aromatic acyl chlorides gave the expected protected bis-amides monomers with good yields. Optimization of the addition reaction was realized by changing the nature of the solvent and the addition mode. Playing on the sterical hindrance of the base such as collidine or lutidine allowed to achieve the synthesis via the unprotected aminoalcohols and to obtain selectively the corresponding bis-amides diols with good yield without side products in two steps. The obtained monomers were fully characterized in details by 1D and 2D NMR correlations experiments to ascertain all the signals, elemental analysis, and mass spectroscopy to confirm the purity. This novel class of biosourced mix bis_amide-diols was the target of a polymerization in bulk to obtain poly(ether)ester-amide. An amorphous polymer (Tg = 92 °C) was obtained as showed by DSC proving the possibility to tune their thermal properties compared to previously reported works. The use of several acyl chlorides with the development of new type of building blocks, in order to control easily the physical and chemical properties of the obtained materials, is undergoing on our laboratory.
Supplementary data
The supplementary material for this paper is available online at http://dx.doi.10.1080/14786419.2015.1124317.
Supplementary_Material.pdf
Download PDF (540.7 KB)Acknowledgments
Authors want to thank all INRAP staff for collaboration especially Mrs Ons Kesraoui and Chaouki Belgacem for mass spectroscopy analysis and M. Mezni for NMR experiments. Authors also want to thank Mrs Fernande Boisson for helpful discussions.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
The financial support of this work by the Ministry of Higher Education, Scientific Research and Information and communication Technologies, Tunisia, is gratefully acknowledged.
References
- Paspirgelyte R, Grazulevicius JV, Grigalevicius S, et al. N,N′-Diphenyl-1,4-phenylenediamine-based enamines containing reactive functional groups as building blocks for electro-active polymers. Des. Monomers Polym. 2009;12:579–587.10.1163/138577209X12519685854626
- Gella IM, Drushlyak TG, Babak NL, et al. Structural analysis of chiral dopants in nematic systems by example of ether–ester-substituted 1,4:3,6-dianhydrohexitols. Mol. Cryst. Liq. Cryst. 2014;591:34–44.10.1080/15421406.2013.833476
- Gomurashvili Z, Kricheldorf HR, Katsarava R. Amino acids based bioanalogous polymers. Synthesis and study of new poly(ester amide)s composed of hydrophobic ɑ-amino acids and dianhydrohexitoles. J. Macromol. Sci. Part A. 2000;37:215–227.10.1081/MA-100101089
- Fenouillot F, Rousseau A, Colomines G, et al. Polymers from renewable 1,4:3,6-dianhydrohexitols (isosorbide, isomannide and isoidide): a review. Prog. Polym. Sci. 2010;35:578–622.10.1016/j.progpolymsci.2009.10.001
- Belgacem C, Medimagh R, Fildier A, et al. Synthesis and characterization of isosorbide-based α,ω-dianhydroxyethersulfone oligomers. Des. Monomers Polym. 2015;18: 64–72.10.1080/15685551.2014.947554
- Bennour H, Fildier A, Chatti S, et al. Biosourced cyclic and multicyclic polyesters based on 1,4:3,6-dianhydrohexitols: application to metal ions uptake in aqueous media. Macromol. Chem. Phys. 2015;216:1081–1090.10.1002/macp.v216.10
- Wu J, Eduard P, Jasinska-Walc L, et al. Fully Isohexide-based polyesters: synthesis, characterization, and structure−properties relations. Macromolecules. 2013;46:384–394.10.1021/ma302209f
- Thiyagarajan S, Gootjes L, Vogelzang W, et al. Chiral building blocks from biomass: 2,5-diamino-2,5-dideoxy-1,4-3,6-dianhydroiditol. Tetrahedron. 2011;67:383–389.10.1016/j.tet.2010.11.031
- Medimagh R, Meddeb Smaidia M, Bennour H, et al. Synthesis and evaluation of the thermal properties of biosourced poly(ether)ureas and copoly(ether)ureas from 1,4:3,6-dianhydrohexitols. Polym. Int. 2014;64:513–520.
- Wu J, Eduard P, Thiyagarajan S, et al. Isohexide derivatives from renewable resources as chiral building blocks. ChemSusChem. 2011;4:599–603.10.1002/cssc.v4.5
- Rose M, Palkovits R. Isosorbide as a renewable platform chemical for versatile applications-quo vadis? ChemSusChem. 2012;5:167–176.10.1002/cssc.201100580
- Feng X, East Anthony J, Hammond W, et al. Sugar-based chemicals for environmentally sustainable applications. In: Korugic-Karasz, L , editor. Contemporary science of polymeric materials. Washington (DC): ACS Symposium Series; American Chemical Society; 2010. p. 3–27.
- Pion F, Ducrot P-H, Allais F. Renewable alternating aliphatic-aromatic copolyesters derived from biobased ferulic acid, diols, and diacids: sustainable polymers with tunable thermal properties. Macromol. Chem. Phys. 2014;215:431–439.10.1002/macp.v215.5
- Chrysanthos M, Galy J, Pascault J-P. Preparation and properties of bio-based epoxy networks derived from isosorbide diglycidyl ether. Polymer. 2011;52:3611–3620.10.1016/j.polymer.2011.06.001
- Caouthar A, Roger P, Tessier M, et al. Synthesis and characterization of new polyamides derived from di(4-cyanophenyl)isosorbide. Eur. Polymer J. 2007;43:220–230.10.1016/j.eurpolymj.2006.08.012
- Caouthar A, Loupy A, Bortolussi M, et al. Synthesis and characterization of new polyamides based on diphenylaminoisosorbide. J. Polym. Sci., Part A: Polym. Chem. 2005;43:2480–2491.10.1002/(ISSN)1099-0518
- Medimagh R, Mghirbi S, Saadaoui A, et al. Synthesis of biosourced polyether-amides from 1,4-3,6-dianhydrohexitols: characterization by NMR and MALDI–TOF mass spectrometry. C.R. Chim. 2013;16:1127–1139.10.1016/j.crci.2013.05.004
- Wu J, Eduard P, Thiyagarajan S, et al. Semicrystalline polyesters based on a novel renewable building block. Macromolecules. 2012;45:5069–5080.10.1021/ma300782h
- Thiem J, Lüders H. Synthesis and directed polycondensation of starch-derived anhydroalditol building units. Starch – Stärke. 1984;36:170–176.10.1002/(ISSN)1521-379X
- Feng X, East Anthony J, Hammond W, et al. Overview of advances in sugar-based polymers. Polym. Adv. Technol. 2011;22:139–150.10.1002/pat.v22.1
- Chatti S, Bortolussi M, Loupy A. Synthesis of new diols derived from dianhydrohexitols ethers under microwave-assisted phase transfer catalysis. Tetrahedron. 2000;56:5877–5883.10.1016/S0040-4020(00)00539-1
- Marín R, Alla A, Martínez de Ilarduya A, et al. Carbohydrate-based polyurethanes: a comparative study of polymers made from isosorbide and 1,4-butanediol. J. Appl. Polym. Sci. 2012;123:986–994.10.1002/app.34545
- Medimagh R, Saadaoui A, Mghirbi S, et al. New biosourced alternated poly(ether)ester-amides (PeEA): synthesis and combined NMR/MALDI TOF MS characterization. J. Polym. Res. 2014;21:486–497.10.1007/s10965-014-0486-4
- Abenhaïm D, Loupy A, Munnier L, et al. Selective alkylations of 1,4:3,6-dianhydro-d-glucitol (isosorbide). Carbohydr. Res. 1994;261:255–266.10.1016/0008-6215(94)84022-9
- Trost B. Comprehensive organic synthesis: reduction. 1st ed. Oxford (UK): Elsevier; 1991.
- Hopton FJ, Thomas GHS. Conformations of some dianhydrohexitols. Can. J. Chem. 1969;47:2395–2401.10.1139/v69-391
- Liu Y, Bhandari B, Zhou W. Glass transition and enthalpy relaxation of amorphous food saccharides: a review. J. Agric. Food. Chem. 2006;54:5701–5717.10.1021/jf060188r