
Abstract
The mechanical properties of biocompatible poly(L-lactide) (PLA) could be dramatically enhanced by cross-linking and interesting network structures might be achieved via the formation of branched and star-shaped structures prior to such cross-linking. However, the synthesis of a four-armed star-shaped PLA is limited to bulk melt polymerization at relatively high temperatures that may degrade the monomer and render the reaction kinetics difficult to control. In this work, a solution-based polymerization approach is introduced with dimethylformamide as the solvent for the initiator and all other reactants. Varying the reactant concentrations and reaction temperature, four-arm PLA star structures were produced in solution and their properties were analyzed. The progress of the polymerization reaction was studied using the 1H NMR and Fourier transform infrared spectroscopy. The quantitative evaluation of the monomer conversion was discussed on theoretical basis. The thermal properties and the crystallinity of the product polymers were evaluated using differential scanning calorimetry. Results indicate polymerization and cross-linking of the four-armed PLA were successful. The introduced solution-based method could open venues for better controlled and more economic reactions of star-shaped and cross-linked PLA.
1. Introduction
Poly(L-lactide) (PLA) is a biocompatible and bioresorbable polyester that is widely used in biomedical applications [Citation1] as a homopolymer or in a mix with other polymers. These biomedical applications extend from orthopedic fixation devices,[Citation2] wound resorbable sutures,[Citation3] artificial skin,[Citation4] stents,[Citation5] controlled drug delivery,[Citation6] to tissue engineering.[Citation7] Polymerization of PLA can basically be achieved by two ways: polycondensation (step-growth) reaction of lactic acid, and ring-opening polymerization (ROP) of lactide monomers.[Citation8] During step-growth polymerization, challenges include the removal of water as a side-product. This type of polymerization leads to products with low molecular weight.[Citation9,10] Therefore, ROP of lactide monomers, requiring no water removal and yielding high-molecular weight PLA, has clear advantages.[Citation8] The driving force for ROP is the relief of conformation strains in the cyclic monomers and thus thermodynamically favorable as it results in reducing the total free energy of the molecule.[Citation11] For ROP to be successful, a catalyst is required. Coordination–insertion mechanism can then take place.[Citation1,9,10] The catalysts usually used for these purposes are organometallic catalysts with zinc (II), titanium (IV), aluminum (III), and tin (II) as shown in Figure (a).
Figure 1. (a) Structure of organometallic catalysts: tin(II) octanoate [Sn(Oct)2], aluminum(III) isopropoxide [Al(Oi–Pr)3], and zinc(II) lactate [Zn(Lact)2]. (b) Structure of organocatalysts.
![Figure 1. (a) Structure of organometallic catalysts: tin(II) octanoate [Sn(Oct)2], aluminum(III) isopropoxide [Al(Oi–Pr)3], and zinc(II) lactate [Zn(Lact)2]. (b) Structure of organocatalysts.](/cms/asset/875fec44-d9a1-4409-92dc-321ee3954b65/tdmp_a_1136532_f0001_b.gif)
It is essential for biomedical applications of PLA to remove all traces of the metallic elements left behind from the catalyst since they may have serious effects on the properties of the polymeric product as well as its toxicity. For example, tin (commonly used in ROP of lactides) leaves residues in ROP products at a concentration higher than 300 ppm which can have a toxic effect in biomedical devices.[Citation1] Therefore, organocatalysts (Figure (b)) were introduced to replace the conventional organometallic catalysts in ROP.[Citation12] Examples of these catalysts include 4-(dimethylamino)pyridine (DMAP), N-heterocyclic carbene, and thiourea-amine (TA). These catalysts proved to be effective in leading to high-molecular weight living polymers at high conversion rates.[Citation10,13,14] Kadota et al. [Citation15] used DMAP together with its protonated form (DMAP.HX) as organocatalysts with both basic and acidic sites. This combination was more effective than DMAP alone in ROP of PLA.
ROP also requires an initiator besides the catalyst for the reaction to proceed. Nucleophilic initiators are selected for this purpose where the number of active sites of the initiator determines the architecture of the product polymers. Nucleophiles with one active site (e.g. ethanol) result in linear PLA [Citation11,12,16], while using polyol compounds (e.g. pentaerythritol) yields branched chains of four-arm star-shaped PLA.[Citation1,14,17,18]
Common methods for PLA formation through ROP are bulk and solution methods. Synthesis in solution is mainly applied if production of linear PLA at low temperatures is desired [Citation19], while bulk polymerization is commonly used to obtain branched PLA. The reason for solution polymerization not to be used for most PLA branched architectures is the low solubility of the initiators. These initiators are usually highly polar, have high melting points, and do not easily dissolve in common solvents together with lactide monomer and polymers.[Citation20]
On the other hand, three major problems are typically encountered with bulk polymerization of branched and star-shaped PLA. First, the reacting monomer, initiator, and catalyst as well as the produced polymer will simultaneously be present in the bulk and the polymer might be soluble in the monomer. Therefore, the viscosity of the reaction mixture increases with time as more polymer is produced. Due to diffusion limitations,[Citation21] it becomes difficult to remove traces of non-reacted components after polymerization. Second, the reaction rates in bulk polymerization are not easily controlled because of the heat of polymerization and the development of hot spots may lead to the auto-acceleration effects.[Citation21,22] Third, for the currently reported systems for bulk polymerization of branched PLA, temperatures in the range between 130 °C and, more frequently, 180 °C have been applied. These temperatures may not be suitable for processing lactide monomer which might evaporate even before these elevated temperatures are reached. The boiling point of the monomer is around 140 °C.
The above described challenges of bulk polymerization can be overcome using a solution polymerization approach at relatively low temperatures and with better thermal control over the reaction. The heat of polymerization would be dissipated through the greater mass of the solvent and the viscosity increase limited.[Citation21] Thus, the main focus of this work is to introduce such solution-based method for polymerizing four-arm PLA and simultaneously handle the lack of solubility of the polyol initiator (pentaerythritol) in a common solvent for all reactants (monomer and catalyst). After forming a star-shaped PLA, cross-linking is performed to reinforce the material for improved mechanical stability.
2. Materials and methods
2.1. Materials
(3S)-cis-3,6-dimethyl-1,4-dioxane-2,5-dione (L-lactide monomer) with a purity of 98%, 4-DMAP catalyst with a purity of ≥99%, 2,2-bis(hydroxymethyl) 1,3-propanediol (pentaerythritol), and hexamethylene diisocyanate (HDI) were purchased from Sigma–Aldrich. Dimethylformamide (DMF) was purchased from Fisher Scientific and used as a solvent during the synthesis of the branched polymers. Dichloromethane (DCM) was used for the extraction. DCM and stabilized tetrahydrofuran (THF), for use in gel chromatography, were obtained from Mallinckrodt Baker Inc. For the characterization, a commercial sample of PLA (purchased from Druect Corp., Pelham, AL) was used as a reference.
2.2. Synthesis
2.2.1. Synthesis of the star-shaped PLA
L-lactide monomer was dissolved in DMF together with the catalyst and the pentaerythritol initiator in a well-sealed round-bottom flask. The reaction took place at a constant temperature (60 °C) in an oil bath. DMF was dried from water residues by mixing it with anhydrous sodium sulfate and passing it through 3 Å molecular sieves at least 24 h before use. The experimental details of the reactions can be found in Table , and the schematic representation for the reaction is illustrated in Figure .
Table 1. Reaction conditions to synthesize PLA.
Figure 2. Schematic representation for the synthesis of branched PLA.
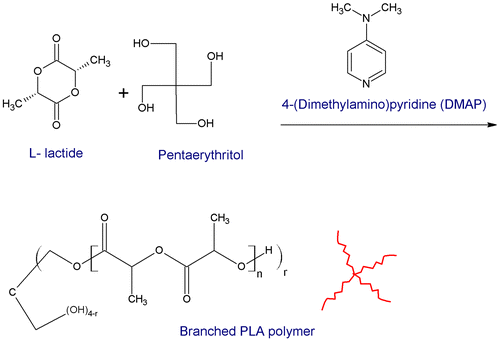
The reactants were added at different ratios of initiator : catalyst : monomer as indicated in Table . Half of the samples were prepared at low catalyst concentrations, that is 4 : 1 equivalent as measured by the ratio of amines in the catalyst to the hydroxyl groups in the initiator. The remaining samples were prepared at higher catalyst concentrations to enhance the kinetics of the reaction.[Citation12]
For all samples, the solution of the produced polymer was extracted using 0.5 N HCl and DCM as the organic phase. The polymer was extracted with DCM from the aqueous layer three times, then the extracted organic solution was washed three more times with deionized water. To facilitate the extraction and avoid the formation of an emulsion, a small amount of a brine solution was added. Excess amounts of water were added to the solution during the extraction to remove DMF which is miscible in water. The collected organic phase was dried with magnesium sulfate followed by filtration under vacuum. The obtained transparent solution was placed in the oven at 60 °C to evaporate the solvent and collect the polymer product.
2.2.2. PLA cross-linking
The cross-linking of PLA was performed using HDI as a cross-linker; 5 μL were added with a DMAP catalyst to the reactor. The reactor was sealed and equilibrated at 35 °C for 5 hours with stirring. To monitor the reaction, a few drops of the solution were placed on a special polyethylene card for Fourier transform infrared spectroscopy (FTIR) analysis. The card was scanned as a background at the beginning of the reaction, and subsequently scanned at time intervals of 3 min during the initial phases of the reaction, and every 10 min after about 40 min.
2.3. Characterization
2.3.1. Fourier transform infrared spectroscopy (FTIR)
Samples were evaluated with Nicolet 6700 FT-IR spectrometer (Thermo Electron Corp., Madison, WI) with a KBr beam-splitter using attenuated total reflectance sampling mode. The spectra were processed using OMNIC 7.3 software package. Results were recorded for the samples with 98 scans, a resolution of 4 cm−1, and data spacing of 1.928 cm−1.
For the quantization of the lactide monomer amount in the product of the polymerization reaction and its ratio to PLA as a main and required product, Beer’s law can be implemented with the aid of infrared spectroscopy. According to Beer’s law,[Citation23] the absorbance of the infrared light at a specific wavelength a(λ) is proportional to the product of the concentration (c) of the vibrating bond in the sample (mol L−1), and the sample thickness l (cm) through a material constant that represents the molar absorptivity e(λ) (L cm−1 mol−1). This relation can be stated as shown in Equation Equation1(1) :
(1)
Deviations from the linear relationship (Equation (Equation1(1) )) can be observed at high concentrations of the absorbing bond and in highly scattering materials (here even at lower bond concentrations).[Citation23] It is important to eliminate the dependence of absolute concentration on infrared absorption, which is possible in binary mixtures if the intensity of a characteristic wavelength (
) for one component can be normalized by the signal at a wave number (
) shared by both compounds.
In the case of the binary mixture of PLA polymer and lactide monomer, the amount of the polymer in this mixture can be quantized by the ratio of absorbance (ZP/M) at the characteristic wavelength to the absorbance at the shared wavelength
modifying Equation (Equation1
(1) ) to obtain Equation (Equation2
(2) ).
(2)
Here, is the molar concentration of the functional groups (chemical bonds) characterized by the wavelength
in the monomer (M) structure;
, the molar absorptivity of the monomer at the wave number
;
, the molar concentration of the functional groups at the wavelength
in the polymer (P);
, the molar absorptivity of the polymer at the wave number
;
, the molar concentration of the monomer based on the bonds at the wavelength
; and
, the molar absorptivity of the monomer at the wave number
.
The molar concentrations of the components can be expressed in terms of weight fraction of the components (wM and wP) and the total concentration (cT) (Equation (Equation3(3) )).
(3)
However, the conversion X of the polymerization reaction is defined as shown in Equations (Equation4(4) ) and (Equation5
(5) ).
(4)
(5)
Simplifying Equation (Equation3(3) ) leads to Equation (Equation6
(6) ) and substituting with Equation (Equation5
(5) ), Equation (7) is obtained.
(6)
(7)
Equation (Equation7(7) ) can be rearranged to give the relation shown in Equation (Equation8
(8) ).
(8)
Equation (Equation8(8) ) implies that there is a linear relationship between the inverse of the polymer’s absorbance ratio in the mixture (
and the amount that represents the conversion (
). This linear relation can be exploited for the calculation of the calibration curve.
2.3.2. Nuclear magnetic resonance (NMR)
1H NMR was performed using 400 MHz Varian NMR spectrometer (Varian Inc., Palo Alto, CA) and data were analyzed using ACD/NMR processor (Advanced Chemistry Development Inc., Toronto, Ontario – Canada). Polymer samples were dissolved in deuterated chloroform (CDCl3) at 26 °C and the chemical shifts (δ) were expressed with respect to the signal of tetramethylsilane as an internal reference. Both lactide and pentaerythritol were dissolved and tested in deuterated dimethyl sulfoxide (DMSO).
2.3.3. Thermal gravimetric analysis (TGA)
Thermal gravimetric analysis (TGA) was performed using a Q500 TGA (TA Instruments, New Castle, DE). Samples were placed in platinum pans and purged with nitrogen at a rate of 60.0 mL/min. The instrument was calibrated using calcium oxalate monohydrate standard following the procedure provided by the instrument manufacturer. Samples were tested by recording the mass change with the temperature increase to 500 °C at a rate of 10 °C/min. The results were plotted and analyzed using TA Instruments Universal Analysis package (Version 4.5A).
2.3.4. Differential scanning calorimetry (DSC)
Thermal transitions were evaluated using a Q2000 Differential Scanning Calorimeter (TA Instruments, New Castle, DE). Samples were placed in standard aluminum pans under a nitrogen environment at a flow rate of 50.0 mL/min. The temperature of the sample was decreased to −5 °C at a rate of 10 °C/min then ramped up to 200 °C at the same rate. The procedure was repeated two times for each sample. The results were analyzed using TA Instruments Universal Analysis package (Version 4.5A).
2.3.5. Molecular weight determination
Molecular weights of the polymeric samples were measured by the Dionex™ high-performance liquid chromatography (UHPLC) system (Thermo Scientific, UltiMate® 3000 HPLC series) equipped with Phenomenex Phenogel™ column. The column is 30 cm long, 7.8 mm internal diameter, 1 μm pore size, 5 μm particle size, solid supported with cross-linked styrene–divinyl–benzene, working at a temperature of 35 °C, and recommended for the 5–500 K Dalton molecular weights. The instrument was installed with the Dionex™ UltiMate™ 3000 rapid separation diode array detector (for UV–vis detection), as well as a Varian 385-LC evaporative light scattering detector (ELSD). The ELSD works with a 480-nm LED light source and was set at 80 °C as evaporating temperature and 90 °C as nebulizer temperature. The gas flow rate was set at 1.5 L/min and the light intensity at 10%. The system was calibrated using standard polystyrene samples with a molecular weight that ranges between 12.5 and 96 K Dalton (purchased from Phenomenex, Czech Republic). PLA samples were dissolved in HPLC grade THF (from Chromservis Prague, Czech republic) at concentrations in the range of 8.8–19.05 mg/mL and filtered during injection with 0.22 μm PTFE membrane filter. The flow rate of the mobile phase in the system was 1 mL/min with 20 μL injected samples that were recorded at 220 nm for 14 min.
3. Results and discussion
The four-arm star-shaped PLA was synthesized according to the core-first approach [Citation24] which allows for better control of the structure and length for each arm than the arm-first approach.[Citation25] In the core-first approach, polymer chains grow from each active end group of the initiator; thus, the number of arms in the final polymer is close to the number of functionalized end groups in the initiator. The monomer-activated mechanism [Citation12] was suggested for the ROP reaction where the nucleophilic attack takes place between the hydroxyl group and the monomer–DMAP complex. The reaction proceeds when the terminal hydroxyl of the opened dimer acts as a nucleophilic center that can react with another monomer complex. This mechanism has the features of living polymerization reactions where the polymer chains continue to grow as long as monomers are available.[Citation9]
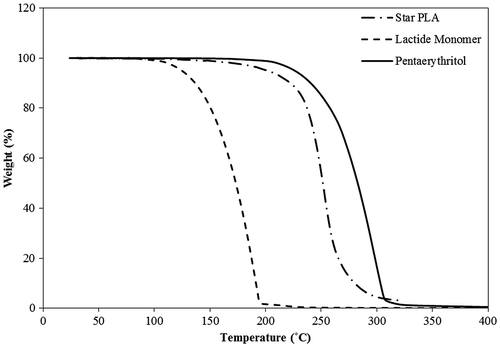
During preliminary experiments, an attempt was made to synthesize the star-shaped PLA using bulk polymerization at different temperatures between 130 and 180 °C. The lactide monomer, however, was found to evaporate and decompose leaving only black residue. With the help of TGA, an early weight loss of about 10% for lactide was observed at a temperature of 120 °C which is the monomer’s melting temperature as given by the provider. The TGA results (Figure ) also showed an excessive loss of monomer at a temperature of 180 °C which can be attributed to the evaporation of the lactide monomer after its boiling point has been reached at around 142 °C. On the other hand, pentaerythritol maintained its structural integrity and did not decompose at elevated temperatures. A weight loss of about 10% for pentaerythritol due to its decomposition was observed only at temperatures of about 250 °C. In traditional bulk polymerization, these high temperatures are required to melt the initiator (pentaerythritol) and allow the progress of the reaction. However, such high temperatures were not suitable for the lactide monomer.
Figure 3. TGA results for the lactide (monomer), the pentaerythritol (initiator), and the produced star-shaped polymer.
Solution-based polymerization was investigated to overcome the challenges of bulk polymerization. The initial focus of this research was on identifying solvents for the reactants, especially the lactide monomer; however, pentaerythritol (the initiator) was difficult to get in solution due to its high polarity and strong hydrogen bonding. DMF was found to dissolve pentaerythritol at temperatures above 50 °C as well as the other reactants and allowed a homogenous solution for the reaction to be formed. The progress of the reaction was followed by 1H NMR analysis of reactants and product polymers.
The monomer showed two characteristic peaks on the 1H NMR spectrum (see Figure ) with the doublet at δ ≈ 1.67 ppm corresponding to the protons on the methyl group and the quartet at δ ≈ 5.10 ppm corresponding to the protons on the adjacent carbon. The results of integration for these peaks matched the number of the six protons on the methyl groups and the two protons bonded to the carbon atoms (labeled 2 and 5 in Figure ).
Figure 4. 1H NMR spectrum for the lactide monomer (top) and pentaerythritol (bottom).
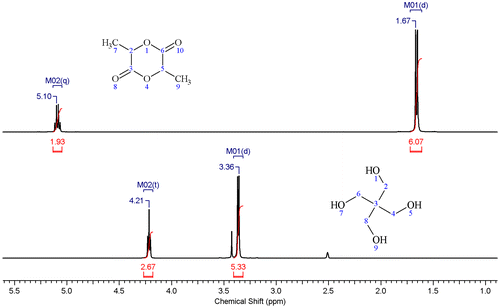
The 1H NMR spectrum for the pentaerythritol (the lower spectrum in Figure ) showed the peaks for the two proton types in the methylene and the hydroxyl groups. The M01 doublet at δ ≈ 3.36 ppm corresponds to the protons in the methylene group, while the M02 triplet at δ ≈ 4.21 ppm corresponds to the protons in the hydroxyl groups. The integration of these peaks resulted in a ratio of 2:1 which is the same ratio of the eight protons in the methylene groups to the four protons in the hydroxyl groups of the pentaerythritol. The other two marked peaks in the spectrum correspond to the DMSO solvent at δ ≈ 2.51 ppm, and water residues at δ ≈ 3.43 ppm.
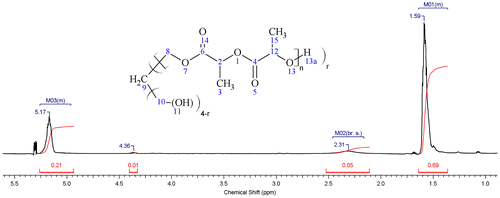
The 1H NMR spectrum of a four-arm star-shaped PLA is shown in Figure . The M01 multiplet at δ ≈ 1.59 ppm corresponds to the methyl pendent groups (labeled 3 and 15 in Figure ); the broad singlet M2 at δ ≈ 2.31 ppm to the hydroxyl end groups (labeled 13); and the M3 multiplet at δ ≈ 5.17 ppm to the hydrogen atoms attached to the alpha carbons (labeled 2 and 12, Figure ). The signal at δ ≈ 4.36 ppm can be assigned to the methylene groups in the pentaerythritol (labels 10, and 8, Figure ). The absence of peaks in the chemical shift region between δ ≈ 3 and 4 indicated the absence of the hydroxyl groups at the ends of the pentaerythritol [Citation26,27] which could imply the growth of a chain arm at each end and the formation of a star-shaped PLA. The molecular weight of the product polymer can be deduced from the NMR spectrum by considering the ratio of PLA to initiator residues resulting in a molecular weight of about 3300 g/mol. This value is comparable to the theoretical molecular weight of about 3693 g/mol calculated for this sample as shown in Table .
Figure 5. 1H NMR spectrum for a star-shaped PLA_H_030 sample (400 MHz, chloroform-d) δ = 4.96−5.26 (7H, m, M03), 2.31 (2H, br. s., M02), 1.36−1.64 (23H, m, M01).
FTIR spectra for the produced four-arm star-shaped PLA samples was investigated and the assignments for the main peaks are reported in Table with a comparison for the assignments reported in the literature.[Citation28] The calculated root mean square error (RMSE) for the experimental and the literature peak vibration values was 3.652 cm−1 which indicated a reasonable matching factor for the experiments.
Table 2. Spectral assignment for linear PLA sample based on the literature [Citation28] compared to the experimental values (calculated RMSE = 3.652 cm−1).
Derivation of the relation between the peaks ratio (ZP/M) and the conversion (X) given in Equation (Equation8(8) ) is universal for binary mixtures. To implement it specifically for the lactide and the PLA, a careful selection is required of characteristic peaks (at wave number λ1) and shared peak (at wave number λ2). Various suggestions can be found in published research. For instance, Messman and Storey [Citation29] chose the peak at 1240 cm−1 which corresponds to the C–O–C bond to show the consumption of the monomer in the reaction. This method, however, requires a non-linear calibration plot for reliable results. Tam et al. [Citation30] considered the peak at 603 cm−1 as it corresponds to the lactide ring deformation. Degee et al. [Citation31] used the peak at 935 cm−1 that corresponds to COO ring breathing mode as the characteristic peak and the shared band at 1383 cm−1 corresponding to the CH3 deformation mode [Citation32] as reference for the calculation of the residual monomer. Braun et al. [Citation23] discussed the peak at 1383 cm−1 and showed that this peak was not isolated, but overlapped with the C–H bending vibration. However, it is sensitive to the polymer chains ordering (crystallization). Braun et al. suggested the peak at 1454 cm−1 that was assigned to the asymmetric bending mode of the C–H3 [Citation33] as a more reliable reference. In our work, a normalization procedure similar to the one discussed by Braun et al. [Citation23] was used, and the characteristic peak λ1 was taken at 935 cm−1, while the shared peak λ2 for normalization was selected at 1454 cm−1. For constructing the calibration curve, the peak area of the band centered at λ1 = 935 cm−1 was calculated between the limits of 946 and 920 cm−1 with the consideration of the baseline established between the points of 967 and 905 cm−1. Similarly, the peak area of the band centered at λ2 = 1454 cm−1 was measured between the limits of 1480 and 1432 cm−1 with the consideration of the straight baseline established between the points of 1500 and 1410 cm−1. The spectral raw data were exported to Matlab® and a code was developed to integrate the curves and calculate the required areas of the assigned peaks.
The calibration curve is shown in Figure . It was established as a straight line using the spectra for pure monomers (with X = 0), pure PLA (X = 1), and pre-defined mixtures. The relation between the conversion term and the inverse of the peak ratio
is shown in Figure with high linearity and a high coefficient of determination (R2 = 0.99) of the data with linear fitting. The observed linearity is in agreement with the theoretical derivation established in Equation (Equation8
(8) ).
Figure 6. Calibration curve for the peaks ratio and their equivalent conversion.
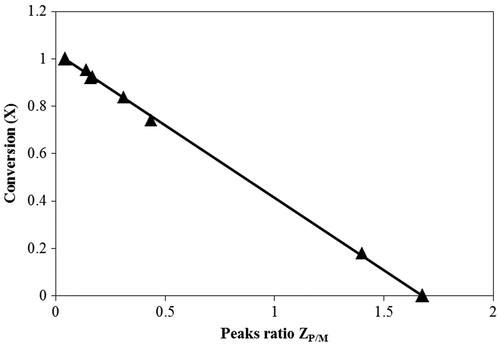
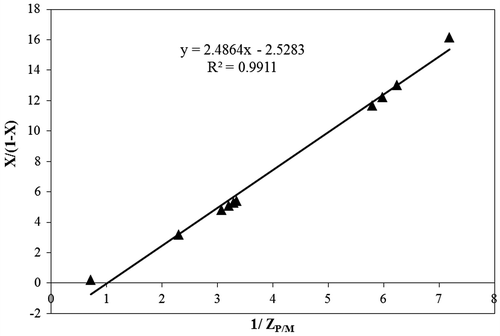
Figure 7. Plot of the experimental data of the conversion term (X/(1 − X)) and the peaks ratio term (1/ZP/M).
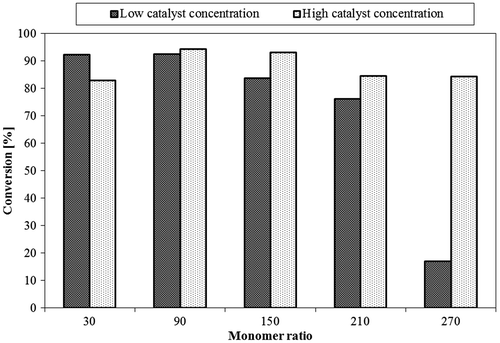
The conversion values for the reactants (Figure ) demonstrated the effect of the catalyst on the reaction. Results showed a consistently high conversion (more than 82%) at higher catalyst concentration, while lower conversion was observed at low catalyst concentration. This might be explained by the steric effect that might occur at high monomer concentration and that can have an effect on conversion. The time of the reaction needed at high catalyst concentration was only about half the time at low concentration which revealed the impact of DMAP on the reaction kinetics. Similar results for slow polymerization at lower catalyst concentration have been reported in the literature.[Citation32]
Figure 8. Effect of different monomer: initiator ratios at high/low catalyst concentrations on conversion.
Thermal properties of the PLA products were evaluated using differential scanning calorimetry (DSC) as shown in Figure . A summery for the transitions is listed in Table . The measured Tg for the samples ranged between 37.75 and 55.6 °C which was within acceptable agreement of the reported range of 50–60 °C for PLA [Citation34]; however, no consistent trend could be found in correlation with the increase in molar ratio of monomer : initiator. This can be understood given that glass transition temperatures mainly depend on the inter-molecular and the intra-molecular forces among the polymer chains and slightly depend on the molecular weight. Melting temperatures increased from 140.82 to 151.63 °C with increasing the ratio and, consequently, with increasing molecular weight of the polymer. These melting temperatures are in agreement with the values reported by Solarski et al. [Citation35] for PLA fibers and generally fall within the Tm range of PLA reported to vary depending on molecular structure and composition.[Citation36]
Figure 9. DSC curves for (1) PLA030, (2) PLA090, (3) PLA150, (4) PLA210, and (5) PLA270 during their second heating cycle.
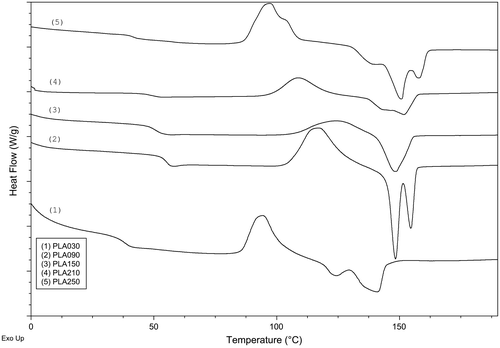
Table 3. Summary of DSC results for PLA linear samples.
Table includes data regarding the energy required for both crystallization and melting which were calculated by integrating the area under the heat flow curves (see Figure ) at the crystallization and the melting temperatures, respectively. Results show a good correlation between these energy values, which could be expected because energy used in the process of melting is actually consumed to melt the crystalline regions in the polymer structure. The higher the crystallinity of a polymer, the higher is the crystallization energy and the melting energy. Melting energy of the polymer is useful in calculating its apparent crystallinity (Xc) by evaluating the ratio of the melting energy (ΔHm) to the enthalpy of fusion for a theoretically perfect crystalline polymer. In the case of PLA (ΔHf), this value amounts to –93.7 J/g.[Citation37] Although the calculated values for crystallinity change between 21.78 and 47.15%, there was no clear trend between this change and the increase in the molecular weight.
It is interesting that most of the PLA products showed a bimodal melting peak (Figure ). This type of melting behavior has been reported for PLA and other semi-crystalline polymers, especially for PLA prepared by solvent evaporation method.[Citation38] Double endothermic peaks were explained by simultaneous melting and recrystallization that took place during melting.[Citation39] Yasuniwa et al. [Citation40] studied the recrystallization by wide-angle X-ray diffraction on poly(butylene naphthalate) and in a similar study on PLA.
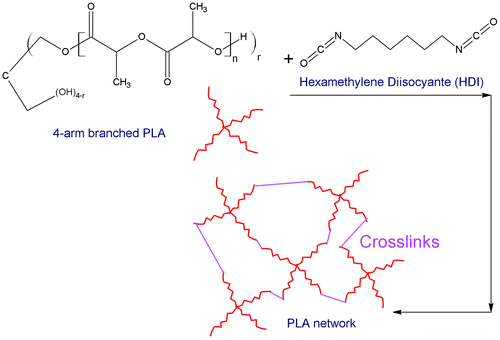
Networked structure of PLA
The obtained four-arm star-shaped PLA was cross-linked by dissolving in methylene chloride and the addition of the HDI cross-linker. HDI reacts with the hydroxyl end groups of the PLA to form urethane bonds. It is expected that a network structure is formed as demonstrated schematically in Figure .
Figure 10. Schematic representation of PLA cross-linking reaction and network formation.
The FTIR spectrum of isocyanate group (N=C=O) vibration falls in the range 2300–2270 cm−1 [Citation41] and, in case of urethane formation, the band at about 2284 cm−1 usually weakens while a new peak around 1700 cm−1 appears.[Citation42] In order to confirm cross-linking of PLA, the reaction was monitored using an FTIR and the depletion of isocyanate was recorded by FTIR at different times. The intensity of the peak at 2280 cm−1 is proportional to the concentration of the HDI cross-linker. Figure shows the behavior of this peak at different times.
Figure 11. Decreasing isocyanate band as monitored over time with FTIR.
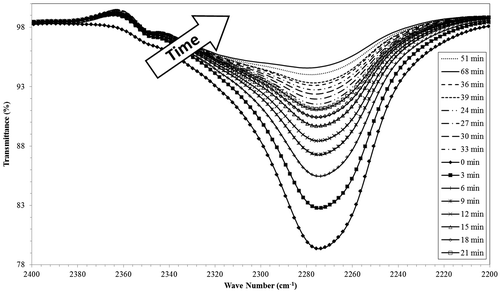
The intensity of the isocyanate peak clearly decreased in a non-linear fashion with reaction time which indicated its reactivity and the potential cross-linking of PLA. An initially rapid decrease was observed that slowed with time. Based on these spectra, the areas under the peaks were integrated and plotted versus time (Figure ). The conversion of the reaction (X) at any time (t) can be calculated according to the relation shown in Equation (Equation9(9) ):
(9)
Figure 12. Change in area under the isocyanate peaks with time.
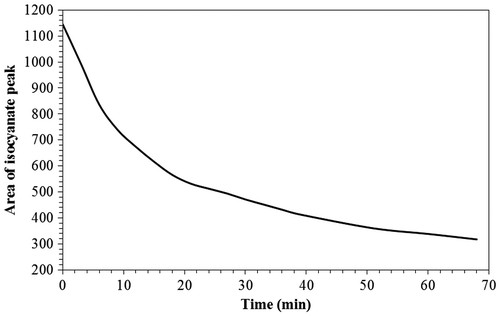
where is the concentration of the HDI cross-linker at the beginning of the reaction (time = 0 min) and CA(t), the concentration of the HDI at a specified time. The calculated conversion after 60 min was 72.25% indicating that the reaction times of a few hours were needed to achieve the required cross-linking.
4. Conclusion
Samples of four-arm star-shaped PLA were produced using solution-based ring opening polymerization in the presence of an organocatalyst. The main challenge for this type of solution polymerization was to identify a common solvent for the pentaerythritol initiator and all other reactants. DMF was found to be suitable to dissolve pentaerythritol at temperatures above 50 °C and simultaneously provide the medium for the solution-based polymerization of the star-shaped PLA. The effect of reactant concentration was studied by modifying the monomer:initiator ratio as well as the catalyst concentration and the reaction time. The development of a star-shaped PLA was evaluated using 1H NMR analysis and the progress of the reaction was followed with FTIR spectroscopy. Conversion of lactide during the reaction was monitored by normalizing the ratio of the lactide monomer ring breathing band at 935 cm−1 to the asymmetric bending of methyl group at 1454 cm−1 which is shared between the monomer and the polymer. Results fitted the linear model that was derived for the relation between the conversion and this peak ratio. Monomer conversion showed consistently high values at higher catalyst concentrations and decreased values at lower concentrations which might be due to the steric effect of monomers at lower catalyst concentrations. The thermal properties of star-shaped PLA were investigated. A semi-crystalline structure was determined by DSC based on the calculation of the apparent crystallinity of the polymer chains. The cross-linking reaction of the star-shaped polymer was monitored by FTIR spectroscopy. Results showed the progress of cross-linking as implied by the depletion of the cross-linker. Although the four-arm star-shaped PLA was directly investigated using 1H NMR and FTIR, other indirect characterization methods might be necessary to further investigate the degree of branching and the length of the formed cross-links as well as to establish their effect on the product properties. The synthesis route explored in this research could open venues for forming cross-linked biopolymer networks that might lead to very useful biomedical applications.
Acknowledgment
The authors are thankful to the effort exerted by Dr. Sherif Hammad for his help with the analysis of the NMR results, and Mr. Vít Novotný of the Nanomaterials institute (CXI) in Liberec, Czech Republic, for performing the HPLC tests.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Albertsson A-C, Varma IK. Recent developments in ring opening polymerization of lactones for biomedical applications. Biomacromolecules. 2003;4:1466–1486.10.1021/bm034247a
- Leenslag JW, Pennings AJ, Bos RRM, et al. Resorbable materials of poly(l-lactide). VI. Plates and screws for internal fracture fixation. Biomaterials. 1987;8:70–73.10.1016/0142-9612(87)90034-2
- Bezwada RS, Jamiolkowski DD, Lee I-Y, et al. Monocryl® suture, a new ultra-pliable absorbable monofilament suture. Biomaterials. 1995;16:1141–1148.10.1016/0142-9612(95)93577-Z
- Gogolewski S, Pennings AJ. An artificial skin based on biodegradable mixtures of polylactides and polyurethanes for full-thickness skin wound covering. Die Makromol. Chemie, Rapid Commun. 1983;4:675–680.10.1002/marc.1983.030041008
- Tamai H, Igaki K, Kyo E, et al. Initial and 6-month results of biodegradable poly-l-lactic acid coronary stents in humans. Circulation. 2000;102:399–404.10.1161/01.CIR.102.4.399
- Buntner B, Nowak M, Kasperczyk J, et al. The application of microspheres from the copolymers of lactide and ϵ-caprolactone to the controlled release of steroids. J. Control. Release. 1998;56:159–167.10.1016/S0168-3659(98)00085-6
- Ishaug SL, Crane GM, Miller MJ, et al. Bone formation by three-dimensional stromal osteoblast culture in biodegradable polymer scaffolds. J. Biomed. Mater. Res. 1997;36:17–28.10.1002/(ISSN)1097-4636
- Kister G, Cassanas G, Fabrègue E, et al. Vibrational analysis of ring-opening polymerizations of glycolide, L-lactide and D,L-lactide. Eur. Polym. J. 1992;28:1273–1277.10.1016/0014-3057(92)90218-Q
- Mehta R, Kumar V, Bhunia H, et al. Synthesis of poly(lactic acid): a review. J. Macromol. Sci. Part C Polym. Rev. 2005;45:325–349.10.1080/15321790500304148
- Jerome C, Lecomte P. Recent advances in the synthesis of aliphatic polyesters by ring-opening polymerization☆. Adv. Drug Deliv. Rev. 2008;60:1056–1076.10.1016/j.addr.2008.02.008
- Odian GG, Odian G Principles of polymerization. vol. 3. New York: Wiley-Interscience; 2004.
- Nederberg F, Connor EF, Möller M, et al. New paradigms for organic catalysts: the first organocatalytic living polymerization. Angew. Chem. Int. Ed. 2001;40:2712–2715.10.1002/(ISSN)1521-3773
- Tsuji H. Poly(lactide) stereocomplexes: formation, structure, properties, degradation, and applications. Macromol. Biosci. 2005;5:569–597.10.1002/(ISSN)1616-5195
- Dechy-Cabaret O, Martin-Vaca B, Bourissou D. Controlled ring-opening polymerization of lactide and glycolide. Chem. Rev. 2004;104:6147–6176.10.1021/cr040002s
- Kadota J, Pavlović D, Desvergne J-P, et al. Ring-opening polymerization of l-lactide catalyzed by an organocatalytic system combining acidic and basic sites. Macromolecules. 2010;43:8874–8879.10.1021/ma101688d
- Connor EF, Nyce GW, Myers M, et al. First example of N-heterocyclic carbenes as catalysts for living polymerization: organocatalytic ring-opening polymerization of cyclic esters. J. Am. Chem. Soc. 2002;124:914–915.10.1021/ja0173324
- Inoue K, Yamashiro M, Iji M. Recyclable shape-memory polymer: poly (lactic acid) crosslinked by a thermoreversible Diels–Alder reaction. J. Appl. Polym. Sci. 2009;112:876–885.10.1002/app.v112:2
- McKee MG, Unal S, Wilkes GL, et al. Branched polyesters: recent advances in synthesis and performance. Prog. Polym. Sci. 2005;30:507–539.10.1016/j.progpolymsci.2005.01.009
- Rasal RM, Janorkar AV, Hirt DE. Poly(lactic acid) modifications. Prog. Polym. Sci. 2010;35:338–356.10.1016/j.progpolymsci.2009.12.003
- Biela T, Duda A, Rode K, et al. Characterization of star-shaped poly(l-lactide)s by liquid chromatography at critical conditions. Polymer. 2003;44:1851–1860.10.1016/S0032-3861(03)00030-2
- Rodriguez F, Cohen C, Ober CK, et al. Principles of polymer systems. Boca Raton, FL: CRC Press; 2014.
- Tulig TJ, Tirrell M. Molecular theory of the Trommsdorff effect. Macromolecules. 1981;14:1501–1511.10.1021/ma50006a070
- Braun B, Dorgan JR, Dec SF. Infrared spectroscopic determination of lactide concentration in polylactide: an improved methodology. Macromolecules. 2006;39:9302–9310. doi:10.1021/ma061922a.
- Knischka R, Lutz PJ, Sunder A, et al. Functional poly (ethylene oxide) multiarm star polymers: core-first synthesis using hyperbranched polyglycerol initiators. Macromolecules. 2000;33:315–320.10.1021/ma991192p
- Roovers J, Zhou LL, Toporowski PM, et al. Regular star polymers with 64 and 128 arms. Models for polymeric micelles. Macromolecules. 1993;26:4324–4331.10.1021/ma00068a039
- Helminen AO, Korhonen H, Seppälä JV. Structure modification and crosslinking of methacrylated polylactide oligomers. J. Appl. Polym. Sci. 2002;86:3616–3624.10.1002/(ISSN)1097-4628
- Numata K, Srivastava RK, Finne-Wistrand A, et al. Branched poly (lactide) synthesized by enzymatic polymerization: effects of molecular branches and stereochemistry on enzymatic degradation and alkaline hydrolysis. Biomacromolecules. 2007;8:3115–3125.10.1021/bm700537x
- Mark JE. Physical properties of polymers handbook. New York: Springer; 2007.
- Messman JM, Storey RF. Real-time monitoring of the ring-opening polymerization ofrac-lactide within situ attenuated total reflectance/Fourier transform infrared spectroscopy with conduit and diamond-composite sensor technology. J. Polym. Sci., Part A: Polym. Chem. 2004;42:6238–6247.10.1002/(ISSN)1099-0518
- Tam CN, Bour P, Keiderling TA. Vibrational Optical Activity of (3 S, 6 S)-3, 6-Dimethyl-1, 4-dioxane-2, 5-dione. J. Am. Chem. Soc. 1996;118:10285–10293.10.1021/ja961677i
- Degée P, Dubois P, Jacobsen S, et al. Beneficial effect of triphenylphosphine on the bulk polymerization of L, L-lactide promoted by 2-ethylhexanoic acid tin (II) salt. J. Polym. Sci., Part A: Polym. Chem. 1999;37:2413–2420. doi:10.1002/(SICI)1099-0518(19990715)37:14<2413:AID-POLA15>3.0.CO;2-#.
- Wang J-L, Wang L, Dong C. Synthesis, crystallization, and morphology of star-shaped poly(ɛ-caprolactone). J. Polym. Sci., Part A: Polym. Chem. 2005;43:5449–5457.10.1002/(ISSN)1099-0518
- Kister G, Cassanas G, Vert M. Effects of morphology, conformation and configuration on the IR and Raman spectra of various poly(lactic acid)s. Polymer. 1998;39:267–273.10.1016/S0032-3861(97)00229-2
- Piorkowska E, Kulinski Z, Galeski A, et al. Plasticization of semicrystalline poly(l-lactide) with poly(propylene glycol). Polymer. 2006;47:7178–7188. doi:10.1016/j.polymer.2006.03.115.
- Solarski S, Ferreira M, Devaux E. Characterization of the thermal properties of PLA fibers by modulated differential scanning calorimetry. Polymer. 2005;46:11187–11192.10.1016/j.polymer.2005.10.027
- Södergård A, Stolt M. Properties of lactic acid based polymers and their correlation with composition. Prog. Polym. Sci. 2002;27:1123–1163. doi:10.1016/S0079-6700(02)00012-6.
- Tasaka F, Miyazaki H, Ohya Y, et al. Synthesis of comb-type biodegradable polylactide through depsipeptide−lactide copolymer containing serine residues. Macromolecules. 1999;32:6386–6389.10.1021/ma990766n
- Nakafuku C, Sakoda M. Melting and crystallization of poly (L-lactic acid) and poly (ethylene oxide) binary mixture. Polym. J. 1993;25:909–917.10.1295/polymj.25.909
- Yasuniwa M, Tsubakihara S, Sugimoto Y, et al. Thermal analysis of the double-melting behavior of poly(L-lactic acid). J. Polym. Sci., Part B: Polym. Phys. 2004;42:25–32.10.1002/(ISSN)1099-0488
- Yasuniwa M, Tsubakihara S, Fujioka T. X-ray and DSC studies on the melt-recrystallization process of poly(butylene naphthalate). Thermochim. Acta. 2003;396:75–78.10.1016/S0040-6031(02)00531-2
- Elwell MJ, Ryan AJ, Grünbauer HJM, et al. In-situ studies of structure development during the reactive processing of model flexible polyurethane foam systems using FT-IR spectroscopy, synchrotron SAXS, and rheology. Macromolecules. 1996;29:2960–2968.10.1021/ma9511208
- Xu L, Li C, Ng KYS. In-situ monitoring of urethane formation by FTIR and raman spectroscopy. J. Phys. Chem. A. 2000;104:3952–3957.10.1021/jp992622g