
Abstract
The polymerization kinetics of acrylamide (AAm) initiated by the photoinitiator DAROCUR2959 (DAR2959) under UV light irradiation were investigated. Variations of polymerization rate with AAm concentration and conversion were studied. Under the present low monomer concentrations the gel effect as well the difusion-controlled termination reactions are expected to be postponed or suppressed. The polymerization rate vs. conversion curve of the photoinduced polymerization of AAm was described by two non-stationary rate intervals and the initial rate maximum. In both intervals the polymerization rate decreased with conversion and the decrease was much more pronounced in the low conversion region. Furthermore, the polymerization rate was observed to decrease with increasing monomer concentration. The polymerization behavior was discussed in terms of the intra and intermolecular chain transfer processes (mainly to backbiting) and the decrease of propagation rate coefficient with the monomer concentration. Furthermore, the chain transfer reactions including backbiting step initiate the formation of less reactive radicals and/or the retardation of the polymerizaton.
1. Introduction
Photopolymerization refers to the process of using light as an energy source to induce the conversion of small unsaturated molecules in the liquid state to solid macromolecules thorough polymerization reactions. Photopolymerization deals with those that are induced by light in the UV, visible to IR spectral region. Upon light excitation, the monomers or oligomers may be solidified by two means: polymerization and crosslinking.[Citation1–3] Although initiating radicals may be produced by various photochemical conversion processes like photoscission, abstraction of intramolecular hydrogen, and electron and proton transfer, the most efficient radical initiators developed so far work via bond cleavage.[Citation2,3]
Water-soluble polymers have received the considerable interest of scientists and engineers working on environmental and industrial problem. Polyacrylamide and its (co)polymers comprise the majority of the market. The properties of final polymers and copolymers are regulated by a broad choice of additives and reaction procedures. Polyacrylamide (PAAm) is a water-soluble high polymer and widely used in water treatment, petroleum, textile, paper-making, metallurgical, and other environment protection fields. Polyacrylamide has found numerous applications as a soil conditioner, in waste water treatment, in the cosmetic, paper, and textile industries, and in the laboratory as a solid support for the separation of proteins by electrophoresis. The association properties of acrylamide with a number of (bio)polymers in aqueous solution have been reported and investigated by a fluorescence quenching method previously used in micelles and lipid bilayers.[Citation4] At pH 7.0, acrylamide partitions between the bulk aqueous phase and the (bio)polymers.
Complex nature of photoinitiated polymerization of acrylamide indicates the different variations of polymerization rates with AAm and photoinitiator concentrations (index describing the changes in the polymerization rate for different monomer concentrations, for example, varies in the range 0.6–1.2 [Citation5]). This deviation from the ideal model was discused in terms of the gel effect, cage effect, primary radical termination, solvation of reactants, … The polymerization of water-soluble acrylates (acrylamide, acrylic acid, …) is very fast and the onset of gel effect appears from ca. 20–30% conversion region. The high viscosity of the reaction system favors the gel effect which is responsible for the strong deviation of radical polymerization of acrylamide from the ideal solution polymerization. In order to depress the gel effect and the deviation from the ideal behavior, the low monomer concentration and low rate of initiation were chosen. Dilatometry as the sensible technique was implemented to follow continuously the slight advance in the polymerization reaction under the very low monomer levels.
The whole polymerization kinetics is based on propagation, termination, and chain transfer events. Among them, intramolecular chain transfer via a six-membered ring, the so-called backbiting step, is reported to play a major role in acrylate polymerizations.[Citation6] Through backbiting, the radical functionality is transferred to the antepenultimate monomer segment. This process is accompanied by the transformation of a reactive secondary propagating radical (SPR) into a less reactive tertiary midchain radical (MCR), which is of higher stability and makes backbiting an enthalpy-driven process (Scheme ).[Citation6]
In our previous studies, the thermally initiated polymerization rate of acrylamide increased with increasing monomer concentration.[Citation3] The similar behavior was also observed in the photoinitiated polymerization of AAm irradiated by the UV light with wavelength 365 nm. In the present study, the wavelength 331 nm and the very low conversion range were utilized. According to our best knowledge, this is the first study of the photopolymerization of acrylamide in the aqueous continuous medium where the rate of polymerization decreased with increasing monomer concentration. The reverse dependence of polymerization rate on the monomer concentration in the very low monomer concentration region rules out all aforementioned effects. The present study addresses chain transfer to polymer backbiting during radical homopolymerization of acrylamide in aqueous solution. Photopolymerization investigations accompanied by dilatometric and dynamic light scattering measurements into AAm polymerizations resulted in pronounced variations in the polymerization rates and PMMA coil sizes. The similarity between AAm and other acrylic monomers suggests that chain transfer events including backbiting may occur (the appearance of branched structured), perhaps at higher temperatures than with acrylates and acrylic acid.[Citation8] The polymers are roughly classified into three categories, i.e., linear, branched, and crosslinked polymers (see Figure ).
Figure 1. Structure of linear (a), branched (b), and network (c) polymers.
2. Experimental
Materials
Commercially available acrylamide (AAm, Fluka) was used as supplied. Extra pure Darocur (Irgacure) 2959 (DAR, IRG) (2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone, C12H16O4, Mw = 224.25 [Citation3]) was used as supplied by Fluka. Doubly distilled water used as a reaction medium was deprived of oxygen by heating to boiling point and cooling under a stream of argon.
Polymerization procedure
The polymerization experiments were performed on an optical bench using UV light of wavelengths λ = 313 nm with intensity Io = 6.5 × 10−7 einstein dm−3 s−1 at 23 °C. Amounts of reactants AAm were varied as shown later. UV–vis absorption spectrum of DAR aqueous solution was measured with a UV–visible spectrometer at room temperature. The extinction coefficient of DAR at 313 nm was estimated to be ca. 300 dm3 mol−1 cm−1. The polymerization technique, conversion determination (a dilatometric technique), and the estimation of polymerization rate, light intensity, number of nanoaggregates and number of radicals per nanoaggregate were the same as described earlier [Citation3,9] and some references mentioned herein. Zeta potential (ζ) and dynamic light scattering (DLS) characterizations of AgNPs or their nanoclusters were performed using a Zetasizer Nano-ZS instrument (Malvern) equipped with a 4.0 mV He–Ne 633 nm laser at a measurement temperature of 25 °C. The sample solution was passed through Puradisc polypropylene FP 30/0.2 μM membrane (GE Healthcare) before measurements, and five acquisitions were made to get a mean value for the measurement.
Molecular mass (Mw) determination of a sample was performed with HPLC Shimadzu apparatus equipped with a differential refractometer RID-6A and a UV–vis detector SPD-10AV using the column HEMA-BIO 1000 (8 mm × 250 mm). A set of dextran standards was used for calibration of the column. The viscosity average molecular weight Mv was calculated according to the Mark–Houwink Equation [Citation10]:
(1)
The Mv was converted into the number average molecular weight Mn [11] according to Equation (3) which is valid for polymer with a Schultz–Flory distribution of the molecular weights (Mw/Mn = 2):(2)
where Γ is a gamma function and α is the exponent in the Mark–Houwink equation. Fort calculation of the average molecular weights of PAAm after substituting the respective values, we obtain [Citation12]:(3)
3. Results and discussion
The conversion kinetics of the radical polymerization was followed by using the dilotometric technique (Figure ). The aqueous phase polymerizations of acrylamide under inert (argon) conditions and the irradiation at λ = 313 nm were investigated. The shape of all conversion curves deviates from the shape typical for the ideal solution polymerizations. Its shape is more typical for the precipitation or dead end polymerization. The conversion curves are concave downward nearly throughout the polymerization. The linear part of conversion curve for the aqueous polymerization of AAm is relatively short or it does not appear at all.
Figure 2. Variation of monomer conversion of photoinduced solution polymerization of AAm with reaction time and AAm concentration. Recipe: 27 g H2O, 0.0515 g DAR, AAm concentrations: (1) 0.2 g (0.74 wt%), (2) 0.3 g (1.11), (3) 0.4 g (1.48) and (4) 0.5 g (1.85).
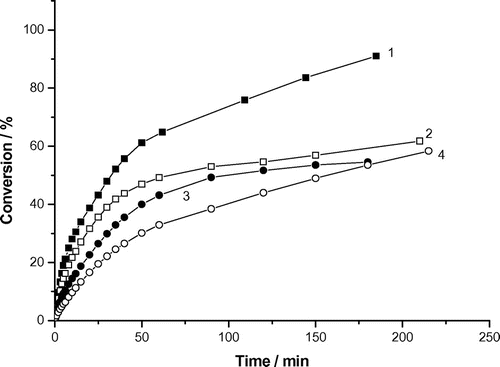
The final conversion (%conv.f within the 3–4 h interval) decreased with increasing monomer concentration as follows;
(4)
This behavior indicates that the final conversion is nearly inversely proportional to the amount of monomer and the polymerization proceeds longer with the smaller amount of monomer (or final polymer) to get similar conversion data. Indeed, the abrupt decrease in the conversion with reaction time data is observed beyond a certain critical polymer concentration (see Figure ) as indicates the following dependence; a critical amount of polymer (g)/ monomer amount (g):(5)
These data indicate that the “limiting” conversion appears beyond the accumulation of the critical amount of polymer (ca. 0.15 g) in the reaction system. Figure shows that the polymerization slows down beyond the “critical” amount of polymer (ca. 0.15 g). The critical conversions ca. 50–90% appear for the runs with 0.5–0.2 g of acrylamide in the monomer feed. These data indicate that the presence of polyAAm is somehow connected with the direct retardation of the polymerization process. They might indicate that the “encapsulation” of radicals within the polymer matrix is responsible for the observed behavior (the formation of less reactive growing radicals by the chain transfer reactions).
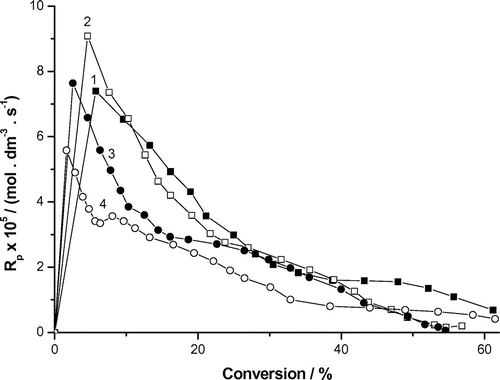
The dependence of the polymerization rate vs. conversion in Figure is described by a curve with two rate intervals (0–10 or 20% (interval 1) and 20–80% conversion (interval 2)) and the one rate maximum at a very low conversion. The extrapolation of the polymerization rates (interval 1) to zero conversion gives the following values of polymerization rate Rp1 × 105 (mol dm−3 s−1) (Figure and Table ):(6)
Figure 3. Variation of the rate of photoinduced polymerization of AAm with conversion and AAm concentrations. Recipe: 27 g H2O, 0.0515 g DAR, AAm concentrations: (1) 0.2 g (0.052 mol dm−3), (2) 0.3 g (0.078 mol dm−3), (3) 0.4 g (0.104 mol dm−3) and (4) 0.5 g (0.26 mol dm−3).
Scheme 1. Backbiting reaction by a [1,5]-H-shift reaction via a six-membered transition state (tagged by *, 2) transferring a secondary propagating chain-end radical (SPR, 1) into a tertiary midchain radical (MCR, 3). The side group is denoted by R, which is a CONH2 ester moiety.[Citation7]
![Scheme 1. Backbiting reaction by a [1,5]-H-shift reaction via a six-membered transition state (tagged by *, 2) transferring a secondary propagating chain-end radical (SPR, 1) into a tertiary midchain radical (MCR, 3). The side group is denoted by R, which is a CONH2 ester moiety.[Citation7]](/cms/asset/7cc20013-10dd-4fae-8dbd-ef47bc1ba791/tdmp_a_1152539_f0004_b.gif)
Table 1. Variation of kinetic and colloidal parameters in the aqueous polymerization of AAm with the AAm concentration.(a)
The experimental data (Figure , Table and Equation (6)) indicate that the polymerization is inversely proportional to the monomer concentration. Futhermore, the abrupt decrease in the polymerization rate with conversion beyond the maximum rate strongly deviates from all kinetic models. The extrapolation of the polymerization rates (interval 2) to zero conversion estimates the value of polymerization rate Rp2 ~ 4 × 105 (mol dm−3 s−1) for all 1–4 runs. The slight decrease in the polymerization rate with conversion agrees more with the trend of the ratio kp/kt obtained for various water-soluble acrylic monomers (acrylic acid, methacrylic acid, acrylamide and methacrylamide) assined to the chain transfer events [Citation7,13] and some references mentioned herein. Herein the polymerization process is accompanied by the transformation of a secondary propagating radical (SPR, kps) into a tertiary midchain radical (MCR, kpt).[Citation6] For these water-soluble monomers an increase of kps towards higher dilution has been reported. This effect was discussed in terms of transition-state theory as being entropy-driven with water as the solvent providing less friction to internal rotational motion of the transition-state structure than the highly dipolar environment in bulk polymerization. For example, with methacrylamide, an increase in kps by ca. 30% has been reported in passing from 20 to 10 wt.% monomer in the aqueous reaction medium. A similar effect has been observed with other monomers such as methacrylic and acrylic acid, N-vinylpyrrolidone and N-vinyl formamide. The transfer from the bulk to diluted solution with methacrylis acid, kps increased at 25 °C even by one order. Above 50 °C, the fraction of MCRs even exceeds the one of SPRs. Under the present experimental conditions (at 23 °C) the fraction of SPRs much exceed the one of MCRs. At 75 °C, kps for 10 wt % AAm is by ca. 8% above kps for 20 wt %, that is, kps ~ 1.5 × 105 dm3 mol−1 s−1 and kpt ~ 45 dm3 mol−1 s−1. Rough extrapolations of literature kp and kt to 23 °C led to kps ~ 2.0 × 104 dm3 mol−1 s−1 and kpt ~ 7.0 dm3 mol−1 s−1, respectively. Figure shows that the polymerization rate slightly decreases with conversion in the interval 2. Indeed the ratio kp/kt obtained for various water-soluble acrylic monomers (acrylic acid, methacrylic acid, acrylamide and methacrylamide) corrected with the chain transfer steps was observed to slightly decrease with conversion [Citation7,13] and some references mentioned herein. The observed decrease in Rp with conversion in interval 2 points out on the increased termination. This might be attributed to backbiting (intramolecular chain transfer) and chain transfer to polymer (intermolecular chain transfer). The transformation of SPRs into less reactive MCR decreases the polymer rate formation. The two types of radicals, SPRs and MCRs, differ distinctly in propagation and termination rate, which results in a strong impact of backbiting on the overall polymerization kinetics.[Citation14] Thus, an increase in MCRs, which accompanies decreased monomer concentration, is associated with a decrease of the effective rate coefficient of propagation, , according to Equation (7).[Citation15]
(7)
However, the abrupt decrease in the polymerization rate with conversion at the very low conversion range indicates that the kp/kts are not able to explain this behavior. Thus, we should include other parameters such as the photoinitiator efficiency, average or local monomer comcentration and impurities. This behavior strongly deviates from the ideal thermally-initiated solution polymerization of acrylic ester monomers which consists of one long stationary state interval and several non-stationary intervals.[Citation16] In our previous paper, the photopolymerization od AAm the polymerization rates increased with increase in the monomer concentration.[Citation3] We have utilized larger concentrations of acrylamide and the polymerization was photoinitiated by UV light with the wavelength λ = 365 nm in the polymerization runs. Furthermore, the dependence of the rate of polymerization vs. conversion was described by a curve with the four rate intervals and two rate maxima, and the maximum rate of polymerization strongly increased with the AAm concentration (Rp,max ~ [AAm]1.4). This was discussed in terms of larger local monomer concentration at the reaction loci (the higher surface activity of AAm, the formation of AAm premicelles and the participation of AAm in the initiation step [Citation17]). A similar behavior was observed also in other radical polymerizations of acrylamide.[Citation1,18] The rate of polymerization in photoinitiated inverse microemulsion polymerization of acrylamide also increased with AAm concentration.[Citation5]
Photopolymerizations of acrylamide carried out under high monomer levels up to high conversions led to the formation of insoluble gells. Furthermore, the chain transfer events also affect polymerizations by an enhanced level of chain branches.[Citation19] It is interesting to note that the light scattering experiments detected the presence of nanogels (crosslinked/branched PAAms) even in the diluted aqueous PAAm solution (Table ). The formation of branched/nanogels during the polymerization process might result from the chain transfer (including backbiting) events. Thus, the nanogels are suggested to take part in the polymerization process, that is, the “encapsulation” of radicals within the polymer matrix might disactivate the reaction loci. Let us further discuss the possible fate of radicals in the nanogels (crosslinked/branched PAAms) according to the procedure applied for the nanoheterogeneous system [Citation20]:(8)
(9)
(10)
where tprop the average time to add one monomer unit into the growing chain in the continuous aqueous phase, tpngel is the average residence time of radicals in the phase before entry into a PAAm microgel, and tter the average time for a termination reaction with another radical in the aqueous phase, NA Avogadro’s number, ka the entry rate coefficient for monomeric radical into the (sub)nanoparticle was taken 4 × 106 dm3 mol−1 s−1,[Citation20] Np the number of nanoaggregates (ca. 1018/dm3), ϕw the volume fraction of aqueous phase (0.98–0.99), kp,AAm the rate constant for AAm propagation (2.4 × 104) dm3 mol−1 s−1 at 23 °C [Citation7,13] and some references mentioned herein, ktss,st the rate constant for termination in the aqueous phase (5.0 × 108 dm3 mol−1 s−1 at 23 °C and [R•.] = 2.7 × 10−5 mol dm−3.[Citation7] By comparing the values of tpngel, tprop, and tter, it was concluded that the propagation and termination events in the aqueous phase are the most probable fate of radicals. Herein, the formation of encapsulated radicals is less pronounced.
The above discussed results show that the the significant lowering of kp toward increasing monomer concentration is dominant effect at medium conversion range. But the variations in the polymerization rates at the low conversion range are too large for being essentially assigned to such kp history. The decrease in kp was also discussed in terms of a decrease in the preexponential factor, A, of kp. The lowering of A (kp) was assigned to intermolecular interactions between the transition state (TS) structure for AAm reaction loci and an AAm environment being significantly stronger that the ones between the TS structure and a water environment. It is speculated that the continuous decrease in Rp with conversion can also be discussed in terms of the intermolecular interactions between the transition state (TS) structure for AAm reaction loci and an AAm environment being weaker that the ones between the TS structure and a PAAm environment.
Among other effects, one can include the local monomer concentration effect (the premicellar structures), impurities (oxygen), and photolysis of reactants might contribute to the whole polymerization process at low conversions. The “local” (a surface active AAm) monomer concentration and its contribution decreases with the degree of conversion. On the contrary Lissi et al. have reported that the fluorescence of fluorescence probes (herein the excited impurities, for example) was quenched by AAm.[Citation21] However, the formation of premicelle AAm/PAAm structures were not experimentally confirmed by any (NMR, spectroscopy, …) measurements.
The synthesis of PAAm was done via an established procedure. Briefly, an aqueous solution of AAm and MI-2959 was irradiated with UVA (313 nm) light (Scheme ).
Scheme 2. Photolysis and initiating radical formation: Darocur (DAR), ketyl radical (KR), benzoyl radical (BR).[Citation22]
![Scheme 2. Photolysis and initiating radical formation: Darocur (DAR), ketyl radical (KR), benzoyl radical (BR).[Citation22]](/cms/asset/3c2947eb-724d-4deb-a840-e4286ee839b2/tdmp_a_1152539_f0005_b.gif)
The reactive benzoyl radicals start the polymerization (the formation of primary and secondary growing radicals) of AAm. The less reactive ketyl radicals (MCR like radicals) mainly take part in termination events. This results from the fact that the activation energy of cross-termination is much smaller that the activation energy of MCR propagation for radical polymerization of acrylate monomers.[Citation23] The reinitiation by KR is not excluded. The radiation of reaction mixture including impurities and oxygen initiate the formation of radicals and the appearance of initial rate maximum. Fasciani et al. have reported that the benzoyl radicals (BRs) under the air conditions are transferred to benzoyl-type acids (BAs).[Citation22] Under the present reaction conditions (with traces of oxygen) the abrupt increase in the initial rate of polymerization might be somehow connected with the formation of peroxides via the excited benzoyl fragments and oxygen.
Table shows the weight, viscosity and number average molecular weights and the molecular weight distribution as a function of AAm concentration. The results obtained show that in spite of the reverse polymerization rate dependence on AAm concentration, the molecular weight of PAAm increases with the monomer concentration. The biradical termination of growing radicals by combination plus disproportionation should lead to values of Mw/Mn below 1.5. The very broad relative molecular weight distribution might result from the strong contribution of the chain transfer events. This behavior might also result from the intramolecular cyclization of PAAm due to which the formation of more densely packed polymer coils appears and Mv is decreased.
4. Conclusion
The stationary UV light homopolymerization of acrylamide (0.74–1.85 wt%) in aqueous solution was investigated by dilatometry, UV spectroscopy and light scattering measurements. Thus, the photopolymerization of acrylamide initiated by the photoinitiator DAROCUR2959 (DAR2959) were studied under the very low monomer concentrations. The polymerization rate vs conversion curve of the photoinduced polymerization of AAm was described by two non-stationary rate intervals and one maximum polymerization rate. The rate of polymerization was observed to decrease with both the concentration of monomer and conversion. This behavior was discussed in terms of chain transfer events (backbiting). It was also confirmed that propagating and termination events in the aqueous phase are most probable fate of radicals. The cross-termination by MCR and ketyl radicals are suggested to take place in the polymerization process.
Disclosure statement
No potential conflict of interest was reported by the author.
Funding
This work was supported by the Agency for the support of science, Slovakia [APVV-0125/11, VEGA-2/0040/14 and -2/0152/13].
References
- Bartoň J, Capek I, Hrdlovič Photoinitiation. II. Kinetics of the acrylonitrule polymerization photoinitiated by aromatic hydrocarbons. J. Polym. Sci. Polym. Chem. 1975;13:2671–2690.
- Fouassier JP. Photoinitiation, photopolymerization and photocuring: fundamentals and applications. Munich, Vienna, New York: Hanser; 1995.
- Capek I. On photoinduced polymerization of acrylamide. Des. Monomers Polym. 2014;17:356–363.
- Blatt E, Chatelier RC, Sawyer WH. Partition and biding constants in micelles and vesicles from fluorescence quenching data. Chem. Phys. Lett. 1984;108:397–400.
- liu l, Yang W. Photoinitiated inverse emulsion polymerization of acrylamide: some mechanistic and kinetic aspects. J. Polym. Sci., Part A: Polym. Chem. 2004;42:846–852.
- Roedel MJ. The molecular structure of polyethylene 1. Chain branching in polyethylene during polymerization. J. Am. Chem. Soc. 1953;75:6110–6112.
- Kattner H, Buback M. EPR study of backbiting in the aqueous-solution polymerization of acrylamide. Macromol. Rapid Commun. 2015;36:2186–2191.
- Seabrook SA, Tonge MP, Gilbert RG. Pulser laser polymerization study of acrylamide in water. J. Polym. Sci., Part A: Polym. Chem. 2005;43:1357–1368.
- Capek I. On photoinduced miniemulsion polymerization of butyl acrylate with clay. Des. Monomers Polym. 2012;15:345–355.
- scholtan w. molekulargewichtsbestimmung von polyacrylamid mittels der ultracentrifuge. Makromol Chem. 1954;14:169–178.
- Vašková V, Juranicova V, Bartoň J. Aqueous polymerization of vinyl monomers in the presence of surfactants. IV. Effect of the chain length of the surfactant on the acrylamide polymerization. Chem.Papers 1986;40:435–441.
- Vašková V, Oremusová D, Bartoň J. Contribution to the study of acrylamide polymerization in the presence of an inhibitor. Makromol. Chem. 1988;189:701–707.
- Kattner H, Buback M. Termination and transfer kinetics of acrylamide homopolymerization in aqueous solution. Macromolecules. 2015;48:7410–7419.
- Scott G E, Senogles E. Polymerization kinetics of n-alkyl acrylates. J. Macromol. Sci., Part A: Chem. 1974;8:753–773.
- Nikitin AN, Hutchinson RA, Buback M, et al. Determination of intramolecular chain transfer and midchain radical propagation rate coefficients for butyl acrylate by pulsed laser polymerization. Macromolecules. 2007;40:8631–8641.
- O’Driscoll KF. An analysis of the factors affecting the pressure dependence of the termination rate constant kt. Die Makromol. Chem. 1979;180:2053–2056.
- Carver MT, Dreyer U, Knoesel R, et al. Kinetics of photopolymerization od acrylamide in AOT reverse micelles. J. Polym. Sci. Part A: Polym. Chem. 1989;27:2161–2177.
- Benda D, Šňupárek J, Čermák V. Inverse emulsion polymerization of acrylamide and salts of acrylic acid. Eur. Poly. J. 1997;33:1345–1352.
- Nikitin AN, Hutchinson RA. Stationary free radical polymerization in the presence of intramolecular transfer to polymer. Macromolecules. 2005;38:1581–1590.
- Guo JS, Sudol ED, Vanderhoff JW, et al. Particle nucleation and monomer partitioning in styrene o/w microemulsion polymerization. J. Polym. Sci. Part A: Polym. Chem. 1992:30:691–702.
- Lissi EA, Encinas MV, Bertolotti SG, et al. Fluorescence quenching of indolic compounds in reverse micelles of AOT. Photochem. Photobiol. 1990;51:53–58.
- Fasciani C, Silvero MJ, Anghel MA, et al. Aspartame-stabilized gold–silver bimetalic biocompatible nanostructures with plasmonic photothermal properties, antibacterial activity, and long-term stability. J. Am. Chem. Soc. 2014;136:17394–17397.
- Barth J, Buback M, Hesse P, et al. Termination and transfer kinetics of butyl acrylate radical polymerization studied via SP-PLP-EPR. Macromolecules. 2010;43:4023–4031.