
Abstract
Organic compounds designed to serve as stable dendrimer cores were developed. A series of aminosugar and amino polyol containing G0 dendrimers were synthesized. The reaction mixture composition was checked by MALDI TOF mass spectrometry, while that of purified products – by 1H and 13C NMR combined with 2D NMR spectroscopy as well as MALDI TOF MSMS mass spectra. Mass spectrometric fragmentation experiments were performed in positive ion mode in order to determine common fragmentation patterns of [M+H]+ ions.
1. Introduction
In recent years, much attention has been paid to targeted specific transport medicines delivery in the body, which is particularly important in the treatment of cancer. As the means for drug molecules transportation, polymer-based nanodevices are applied,[Citation1,2] especially dendrimers.[Citation3] The use of these nanoparticles has several advantages, when compared with conventional therapeutics of low molecular weight, namely, it improves drugs pharmacokinetics, drug penetration through the biological barriers and the therapeutic effect.[Citation4]
Dendrimers as biologically active molecules have been of particular interest in recent decades. Their strongest points are the drug delivery abilities, and plethora of published experimental data available.[Citation5] Yet, drug delivery is only one of many potential applications of dendrimers. Other important dendrimer applications include vaccination systems,[Citation6,7] diagnostic purposes,[Citation8] chemical catalysis [Citation9,10] and chelating agents,[Citation11] but also as stand-alone drugs against Alzheimer’s disease,[Citation12] cancer [Citation13] and other diseases.
Since the properties of dendrimers depend primarily on the composition of the outer shell, the behavior of the dendrimer molecules of similar size, but with a different composition of the coating is different.[Citation14]
Many types of external dendrimer layers have been investigated, of which sugar-based and sugar-mimicking ones are particularly notable. The external surface layers of dendrimers can be made of glycopeptides [Citation7,15–17] (often obtained by automatic synthesis) or sugar amides (obtained from lactone sugar derivatives and amine-terminated dendrimers [Citation18] or from amino sugars or aminoglycosides and carboxy-terminated dendrimers) [Citation19,20]). Another approach uses linkers between sugar moiety and the active site of the dendrimer,[Citation21,22] or even these two components are only electrostatically bonded.[Citation23] However, there still seems to be no data published on binding sugars as a result of a reaction of amino sugar (or other aminopolyol) with epoxide, which was used in experiments described by us below.
Aminopolyols as dendrimer terminal groups might show very interesting properties. Hydroxyl, sugar-derived surface groups ensure excellent water solubility without notable induction of toxicity. Near-surface secondary or tertiary amino moieties provide polycationic nature of dendrimer, which is desirable when interacting with organism cells.[Citation14] If the polyol is sugar based (like the N-methylglucamine used in presented studies), the dendrimer surface might mimic naturally occurring bioparticles of similar size, and interact with sugar-based glycocalyx as they both have almost identical external composition.
As the physicochemical properties of dendrimers depend mainly on the type of terminal groups, located on the surface, the dendrimers of higher generations with the same terminal groups should have similar properties as the dendrimers of generation G0 presented here, irrespectively of internal dendrimer composition. Even though all G0 dendrimers mentioned in this paper differ in the core structure, the molecules show similar solubility, transition metal affinities and metal complexes solubilities, dependent on and similar to those shown by the corresponding unbound aminopolyols.
The aim of this study was to obtain a zero generation dendrimers (G0) having terminal OH groups, useful for functionalization and further development of dendrimer. Reactions of oxirane ring cleavage by nucleophilic functional groups were exploited, thanks to the amine group being more nucleophilic than the hydroxyl moiety. In order to identify the obtained compounds, a selection of spectroscopic methods was used, mainly mass spectrometry and nuclear magnetic resonance. The prepared dendrimers can be further expanded using other glycidylic compounds, which can be opened by OH groups or can be suited to obtain dendritic polyesters in reactions with carboxylic acids or hydroxycarboxylic acids. Further development of the dendrimer may involve the reaction of OH groups with epichlorohydrin without disturbing the oxirane rings, and then repeated cleavage of the epoxide by an aminopolyol. Reactions of this type are used in syntheses of epoxy resins.
2. Experimental
2.1. Abbreviations
Aminopolyol (APO), 2,5-dihydroxybenzoic acid (DHB), Heteronuclear Multiple-Bond Correlation (HMBC), Heteronuclear Single-Quantum Coherence (HSQC), Mass Spectrometry (MS), Matrix-Assisted Laser Desorption/Ionization (MALDI), Nuclear Magnetic Resonance (NMR), methanol (MeOH), % of relative abundance (r.a.), tetramethysilane (TMS), time-of-flight (TOF), tris(2,3-epoxypropyl) isocyanurate or 1,3,5-tris(oxiran-2-ylmethyl)-1,3,5-triazinane-2,4,6-trione or triglycidyl isocyanurate (TGIC), tris(hydroxymethyl) aminomethane (TRIS), two-dimensional nuclear magnetic resonance spectroscopy (2D NMR), zero generation of dendrimer (G0).
2.2. Materials
All reactions were carried out under argon atmosphere. All starting materials, i.e. tris(4-hydroxyphenyl)methane triglycidyl ether, triglycidyl isocyanurate (TGIC), tris(hydroxymethyl)aminomethane (TRIS), 3-methylamino-1,2-propanediol, N-methylglucamine as well as auxiliary materials, 2,5-dihydroxybenzoic acid (MALDI matrix), deuterium oxide (D2O), TMS and methanol were purchased from Sigma-Aldrich (Switzerland) and were used without further purification, except of MeOH, which was dried over molecular sieves. Column chromatography were performed on Macherey-Nagel silica gel 60 (40–63 μm).
2.3. Methods
The structure of the obtained compounds was confirmed by NMR spectroscopy and MALDI mass spectrometry. The 1H and 13C NMR spectra were recorded on a Varian VNMR-S 400 (Varian Inc., Palo Alto, CA, USA) at frequencies 402.643 and 101.254 MHz, respectively, using 5-mm probehead (90° pulse width: 1H = 13.6 μs, 13C = 6.9 μs). Standard one-pulse sequence (s2pul) and acquisition parameters were used. The 13C NMR spectra were recorded with broadband 1H decoupling. The 2D NMR measurements were performed on an Agilent DD2 800 spectrometer (Agilent Technologies, Santa Clara, CA, USA), operating at the frequency of 799.890 MHz for 1H and 201.151 MHz for 13C, equipped with a 1H/13C/15N 5-mm probehead. D2O was used as a solvent, and tetramethylsilane (TMS) as an internal standard. All NMR spectra were acquired at ambient temperature (298 K).
MALDI TOF mass spectra were obtained on a spectrometer Waters/Micromass Q-TOF (time of flight) Premier (Manchester, UK) with a MassLynx V4.1 software, equipped with a 200 Hz repetition rate Nd/YAG laser (l = 355 nm, power density 107 W cm−2), in the positive ion mode using 2,5-dihydroxybenzoic acid (DHB) as a matrix. Samples for MALDI MS were dissolved in methanol (3–5 mg/mL). The obtained solution (20 μL) was mixed with saturated DHB methanolic solution (30 μL), sonicated for 1 min and 1 μL of the resulting solution was deposited on the MALDI target well and left until dryness to prepare the target spots.
All MS/MS experiments were conducted on a Waters/Micromass (Manchester, UK) Q-TOF Premier mass spectrometer, positive ion mode. Argon was used as a collision gas at the flow-rate of 0.5 mL min−1 in the collision cell. Collision energy was held within range of 10/70 eV, with a minimum of four different values per molecule.
2.4. Syntheses
2.4.1. Synthesis of 1–6 – general procedure
Substrates – appropriate triepoxide (I or II) and aminopolyol (III, IV, or V) – were suspended in anhydrous methanol, and the resulting mixture was purged with argon gas. The reaction mixture was heated to 55 °C under argon atmosphere for 60 h. Then, the solvent was evaporated on a rotary evaporator giving a clear, viscous, amorphous product. Compounds 1–3 required purification by column chromatography, the other compounds were used without further purification. The quantities of reactants (in mmol) and volume of solvent (in mL) used in each synthesis are given in Table .
Table 1. The amounts of reactants and solvents used in the synthesis of dendrimers 1–6.
Synthesis of compound 1 (C40H61N3O15): To 3 mmol of tris(4-hydroxyphenyl)methane triglycidyl ether, 15 mmol (5 eq) of tris(hydroxymethyl)methylamine were added. About 50 mL of dried methanol was added and the mixture was rinsed with argon gas. After start-up, the reaction was carried in argon atmosphere and kept at 55 °C for 60 h without waiting for substrates to dissolve. All solids disappeared in the first 2 h of heating. The reaction mixture was then concentrated using a rotatory evaporator. The remaining residue was a yellow, transparent, amorphous solid. Calculated monoisotopic mass: 823.41035, found: 824.4 [M+H]+.
Synthesis of compound 2 (C40H61N3O12): To 7 mmol of tris(4-hydroxyphenyl)methane triglycidyl ether, 21 mmol (3 eq) of 3-methylamino-1,2-propanediol were added. About 30 ml of dried methanol was added and the solution was rinsed with argon gas. After start-up, the reaction was carried out in argon atmosphere and kept at 55 °C for 60 h without waiting for substrates to dissolve. All solids disappeared in the first hour of heating. The reaction resulted in yellowish, transparent, amorphous solid slowly turning opaque and viscous due to its hygroscopic nature. Calculated monoisotopic mass: 775.42559, found: 776.4 [M+H]+.
Synthesis of compound 3 (C49H79N3O21): To 5 mmol of tris(4-hydroxyphenyl)methane triglycidyl ether, 15 mmol (3 eq) of N-methylglucamine were added. About 30 ml of dried methanol was added, the mixture was rinsed with argon gas. After start-up, the reaction was carried out in argon atmosphere and kept at 55 °C for 60 h without waiting for substrates to dissolve. All solids disappeared in the first hour of heating. The reaction gave orange, transparent, amorphous solid slowly turning opaque due to its hygroscopic nature. Calculated monoisotopic mass: 1045.52071, found: 1046.6 [M+H]+.
Synthesis of compound 4 (C24H48N6O15): To 7 mmol tris(2,3-epoxypropyl) isocyanurate 28 mmol (4 eq) of tris(hydroxymethyl)methylamine were added. About 50 ml of dried methanol were added, the mixture was rinsed with argon gas. After start-up, the reaction was carried out in argon atmosphere and kept at 55 °C for 60 h without waiting for the substrates to dissolve. All solids disappeared in the first hour of heating. The workup was limited to solvent evaporation. The reaction gave colorless, transparent, amorphous solid slowly turning opaque due to its hygroscopic nature. Calculated monoisotopic mass: 660.31784, found: 661.3 [M+H]+.
Synthesis of compound 5 (C24H48N6O12): To 7 mmol of tris(2,3-epoxypropyl) isocyanurate, 21 mmol (3 eq) of 3-methylamino-1,2-propanediol were added. About 30 ml of dried methanol was added, the mixture was rinsed with argon gas. After start-up, the reaction was carried out in argon atmosphere and kept at 55 °C for 60 h without waiting for substrates to dissolve. All solids disappeared in the first hour of heating. The workup was limited to solvent evaporation. The reaction gave colorless, transparent, amorphous solid slowly turning opaque and viscous due to its hygroscopic nature. Calculated monoisotopic mass: 612.33308, found: 613.3 [M+H]+.
Synthesis of compound 6 (C33H66N6O21): To 4 mmol of tris(2,3-epoxypropyl) isocyanurate, 12 mmol (3 eq) of N-methylglucamine were added. About 30 ml of dried methanol was added, the mixture was rinsed with argon gas. After start up, the reaction was carried out in argon atmosphere and kept at 55 °C for 60 h without waiting for substrates to dissolve. All solids disappeared in the first half an hour of heating. The workup was limited to solvent evaporation. The reaction resulted in colorless, transparent, amorphous solid slowly turning opaque due to its hygroscopic nature. Calculated monoisotopic mass: 882.42821; found: 883.4 [M+H]+.
3. Results and discussion
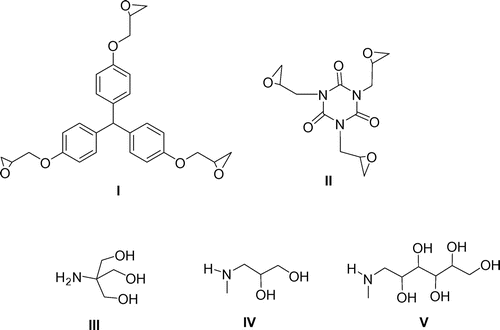
In our experiment, the following groups of compounds were synthesized: compounds 1–3, sharing the same tris(4-hydroxyphenyl)methane central moiety, and compounds 4–6, sharing the same isocyanurate central moiety. All compounds were synthesized in a direct reaction between triglycidyl ethers: I [Tris(4-hydroxyphenyl)methane triglycidyl ether, synonym: triphenylolmethane triglycidyl ether; C28H28O6, molecular weight 460.52] and II [tris(2,3-epoxypropyl) isocyanurate, synonyms: 1,3,5-tris(oxiran-2-ylmethyl)-1,3,5-triazinane-2,4,6-trione, triglycidyl isocyanurate, TGIC; C12H15N3O6, molecular weight: 297.26] and aminopolyols: III – tris(hydroxymethyl)aminomethane (TRIS), IV – 3-methylamino-1,2-propanediol as well as V – N-methylglucamine, respectively (general reaction scheme is shown in Figure ).
Figure 1. General reaction scheme.
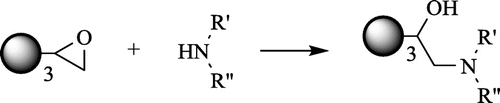
The synthesis was carried out in methanol at 55 °C in the atmosphere of inert gas (argon) according to the preparation procedure described in the section ‘The general procedure of synthesis of 1 – 6.’
The structural formulae of the substrates are shown in Figure .
Figure 2. The structure of the substrates I–V, used for the synthesis of dendrimers 1–6 G0 (I and II – triepoxides, III–V – aminopolyols).
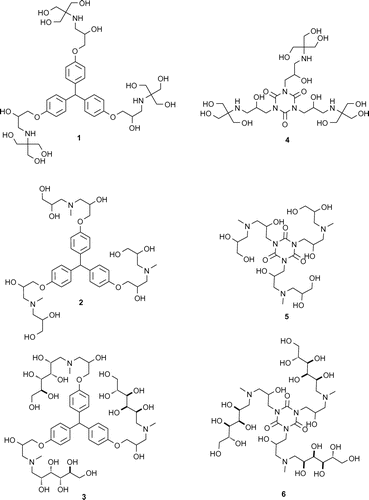
As a result of the reaction, six compounds (1–6) with the structure of dendrimeric polyols were obtained, their formulae are shown in Figure . These compounds contain 12 (compounds 1 and 4), 9 (compounds 2 and 5) and 18 (compounds 3 and 6) terminal hydroxyl groups, which makes them hydrophilic and readily soluble in water. This property is very important for their potential biomedical applications, as they can be used in the natural environment of living organisms.
Figure 3. Compounds obtained (1–6) and used in MS/MS experiment.
3.1. MALDI mass spectrometry
After each reaction the solvent was evaporated and concentrated reaction mixtures containing desired products (1–6, respectively) were subjected to MALDI mass spectrometry.
MALDI mass spectra, measured for the crude compounds, showed the presence of peaks originating from the protonated molecules of the desired products, and also additional peaks of by-products (Figure (a), Table ). Figure (a) shows the MALDI TOF mass spectrum of crude compound 1, as a representative example. It can be seen that besides the main product 1 (A, [M+H]+ m/z 824), there are side products present, in which only two of the three oxirane groups of substrate underwent the reaction and the third remained intact (B) or in which one oxirane ring was opened by an amine group of polyol and the second epoxide ring reacted with solvent i.e. methanol (C) or with water (E). 2,5-Dihydroxybenzoic acid (DHB, MALDI matrix) is also able to react with oxirane in MALDI measurement conditions, competing with other hydroxy compounds in addition reaction to the oxirane ring (D can be a product of addition of one methanol molecule and one DHB molecule to I or it could be a fragment ion derived from B, formed by the loss of C3H4O – Table ). For compounds 4–6, which contain isocyanuric core, only small amounts or traces of by-products were seen in the MALDI spectra (Figure (c)), so these compounds were characterized further without additional purification. Compounds 1–3 required purification using column chromatography with silica gel as stationary phase and chloroform-methanol mixture with increasing MeOH percentage as eluent. After chromatographic purification (Figure (b)), the products were characterized by NMR spectra and selected [M+H]+ ions were submitted to collisions in MALDI MSMS experiment to investigate their fragmentation.
Figure 4. MALDI TOF mass spectrum of: a) a crude compound 1, matrix DHB. Ions: A = [1+H]+ i.e. [I+3Tris+H]+, m/z 824, B = [I+2Tris+H]+, m/z 703; D = [I+DHB+CH3OH+H]+, m/z 647, E =[I+Tris+H2O+H]+., m/z 599; b) purified compound 1, c) crude compound 4.
![Figure 4. MALDI TOF mass spectrum of: a) a crude compound 1, matrix DHB. Ions: A = [1+H]+ i.e. [I+3Tris+H]+, m/z 824, B = [I+2Tris+H]+, m/z 703; D = [I+DHB+CH3OH+H]+, m/z 647, E =[I+Tris+H2O+H]+., m/z 599; b) purified compound 1, c) crude compound 4.](/cms/asset/89e5fc96-d1cc-4af5-85e9-3fe31cc7017e/tdmp_a_1231048_f0004_b.gif)
Table 2. Main ions (m/z, % r.a.) in MALDI TOF positive mass spectra of crude products 1–6 and proposed ion structures; APO = aminopolyol.
The MALDI TOF mass spectrum of 1 revealed the presence of desired addition product of three TRIS molecules to the substrate I (triadduct), proved by the peak [M+H]+ at m/z 823 with relative abundance 100%. There are also admixtures of diadduct [I+2Tris+H]+, peak at m/z 703 50% r.a.), monoadduct with one TRIS molecule and water (peak at m/z 599, 20% r.a.) and, surprisingly, a peak originated from one molecule of DHB and addition of one molecule of methanol to substrate I (m/z 647, 40% r.a.). The latter addition product has to be formed during the mass spectrometric measurement or directly before it, during the sample preparation. In the MALDI mass spectra of 2 and 3, the peaks D are not seen, but the signals of diadduct (B) are more intense (100% r.a.) than those of the desired triadduct A. It should be noted that the relative abundance of a particular peak in the mass spectrum does not bring direct information on the compound concentration in the reaction mixture, because there are many factors which influence the peak abundance, such as ionization efficiency or stability of the protonated molecule and the dependence of relative abundance on concentration is not linear. Concluding, we could not calculate the reaction mixture composition from the mass spectra obtained. However, after chromatographic purification, the additional peaks of side products were absent from the spectrum (Figure (b)) and this fact attests the compound purity. Other proofs of the product purity are the NMR spectra, specifically proton signals integrations.
3.2. NMR characterization
The structures of the obtained compounds were confirmed by 1H and 13C NMR spectroscopy. Compounds 1–3, having a triphenylmethane core, exhibit characteristic signals in the range 6.61–6.85 ppm in 1H NMR spectrum corresponding to the aromatic protons and the signal at 5.41–5.66 ppm assigned to the methine proton in triphenylmethane. For compounds 4–6, there is no proton signal over 5 ppm (Table ).
Table 3. Chemical shifts of proton (1–6) and carbon (2, 4–6) atoms δ (ppm), obtained from NMR spectra, measured in D2O; atom labeling given above the table.
Detailed analysis of 1H and 13C NMR as well as 2D NMR spectra of compound 4 is performed herein as a representative example.
Diastereotopic protons, labeled as d1 and d2 (see the labeling scheme above Table ), give separate signals in the 1H NMR spectrum; d1 – dd (doublet of doublets) at 4.05 ppm and d2 – dq (doublet of quartets) at 3.91 ppm, with coupling constants 13.9 Hz (Jd1,d2, geminal protons), 7.9 Hz (Jd1,e – protons at adjacent carbon atoms) for d1, and Jd2,d1 13.8 Hz, Jd2,e 4.0 Hz for d2, respectively (Figure ). These two protons are situated at the same carbon atom, according to the 1H-13C HSQC (Figure ) spectrum (δ(13C) 49.16 ppm), the same protons correlate through three bonds with the carbon of carbonyl group (C=O, 153.17 ppm) of heterocyclic core, according to 1H-13C HMBC spectrum. The signal originating from proton e appears as a multiplet at 3.98 ppm, since it is coupled with protons d1, d2, f1 and f2.
Figure 5. 1H NMR spectrum of compound 4 (D2O, 298 K).
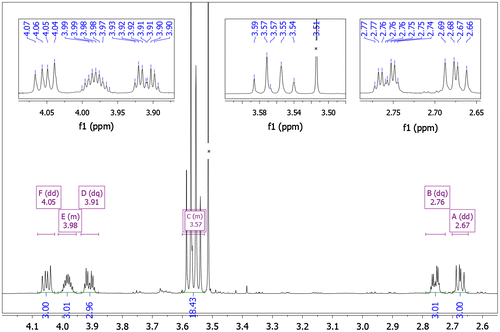
Figure 6. 1H-13C HSQC NMR spectrum of compound 4 (D2O, 298 K) in the range 2.4–4.3 ppm (1H) and 30–180 ppm (13C).
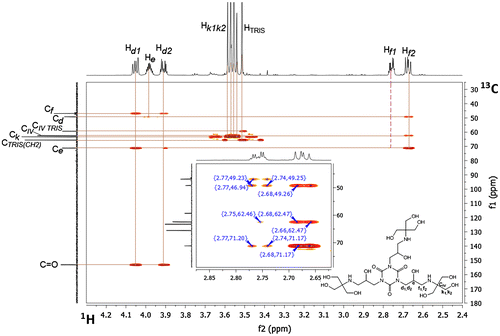
The proton e is attached to the carbon atom C δ 71.17 ppm. The spectral image of 1H NMR signals of diastereotopic protons f1 and f2 looks as almost as a ‘mirror image’ of that of protons d1 and d2: f1 gives doublet of quartets (dq) at 2.76 ppm (J = 11.8 and 3.3 Hz) and f2 gives doublet of doublets (dd) at 2.67 ppm (J = 12.1 and 8.6 Hz). These two protons are situated at the same carbon atom (which in the 13C NMR spectrum gives a signal at δ 46.91 ppm). Integration of the area under each proton peak (d1, d2, e, f1, f2) corresponds to three protons, which confirms complete addition to the three oxirane groups of the substrate, triepoxide II. All protons d1, d2, f1, f2 correlate to the carbon atom at 71.17 ppm through two bonds (HMBC). The carbon atom at δ 62.47 ppm, has no proton attached (HSQC), so it has to correspond to the quaternary carbon atom C(CH2OH)3. There is a correlation between this carbon atom and f1, f2 protons through three bonds noted in the HMBC spectrum (Figure ).
The signals at 3.54, 3.56 and 3.58 ppm (singlets) correspond to the protons of CH2 groups k1 or k2, situated at the same carbon atom δ 63.31 ppm. The proton signal (singlet), situated at 3.51 ppm (not integrated in Figure ), corresponds to the CH2 protons, coming from the residue of TRIS, which was used in excess and was not removed completely after completion of the reaction. From the HSQC spectrum (Figure ) of 4 it could be concluded that these protons are attached to the carbons atoms of chemical shift δ 65.58 ppm, neighboring the carbon atom δ 59.24 ppm. The NMR spectra of neat TRIS., measured separately in D2O, confirmed this observation.
For compounds 1–3, containing methyltriphenyl core, the accurate assignments of carbon atoms is difficult because of many conformational forms of the molecules, since complicated conformational equilibrium is observed even in the 1H NMR spectrum of the unsubstituted substrate and it is getting more complicated after the reaction. Exemplary 13C NMR carbon signal assignments were made only for compound 2 (Table ).
3.3. MALDI TOF MS/MS spectra
To check whether the di- and monoadduct peaks seen in the MALDI mass spectra are not a result of [M+H]+ ions fragmentation, the [M+H]+ ions of compounds 1–6 were chosen as precursor ions and they were fragmented in the collision cell (quadrupole) of MALDI QTOF instrument, using at least four values of collision energy (CE = 15, 30, 50 and 70 eV) for each ion. The aim of this experiment was also to examine the MALDI MS fragmentation patterns, characteristic of protonated molecules of the compounds obtained. It appeared that none of the ions (B–E, Table ) observed in the basic MALDI mass spectra of crude product 1–3 were present in the MSMS spectra of [M+H]+ precursors, so the ions B–E are not collisionally induced fragments.
The [M+H]+ ions of compounds 1–6 are relatively stable in MALDI MS conditions, as the extensive decomposition does not start for collision energies below 50 eV.
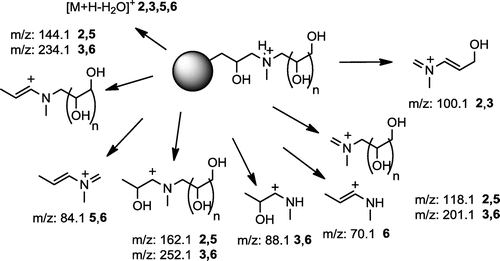
Several common fragmentation patterns were found for the ions analyzed . The simple observation is that for [M+H]+ of compounds 1 and 4, the fragments at m/z 104, 160 and 178 are common, respectively for compounds 2 and 5 the ions at m/z 118, 144 and 162 and for 3 and 6 – at m/z 88, 234 and 252 are common. It is easy to note that there is a close analogy between the most intense ions m/z 178 (14), 162 (25) and 252 (36) – all of them are formed by cleavage of the side chain (substituent) at a heteroatom (O for 1–3, N for 4–6), associated with the molecule core. The proposed structures of the ions at 178, 162 and 252 m/z are shown in Figure :
Figure 7. Proposed structure of most intense fragment ions in MALDI MSMS spectra of 1–6.
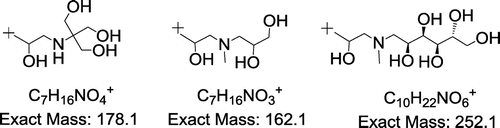
It means that the most intense dissociation is the cleavage of attached aminopolyol moiety together with 2-hydroxypropyl fragment. The cleaved chain retains positive charge and therefore is recorded in the mass spectrum. In turn, the ions at 160, 144 and 234 m/z differ from those at 178, 162 and 252 m/z by 18 mass units (H2O elimination). The ion at m/z 104 is specific of TRIS (III) substituent and appeared in the MSMS spectra of 1 and 4, m/z 118 for polyol IV (MSMS of 2 and 5) and 88 m/z for N-methylglucamine V (MSMS 3 and 6). The full set of fragment ions found in MSMS MALDI QTOF mass spectra of protonated molecules [M+H]+ is presented in Table (positive charge is placed on a randomly selected atom).
Table 4. Full set of fragment ions, found in MSMS MALDI-QTOF experiment of [M+H]+ ions of compounds 1–6.
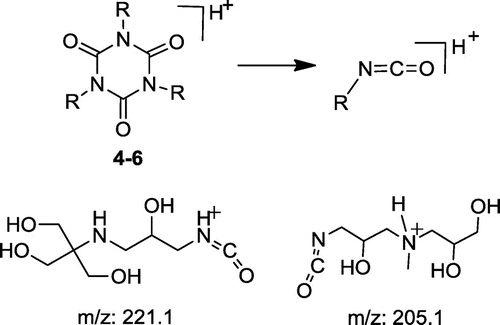
Various decomposition pathways involving isocyanuric center can be observed, including cleavage of the molecule into three equal parts, as depicted in Figure .
The fragments, confirming such a cleavage are those at m/z 221 observed for compound 4 and at m/z 205 in the pattern of compound 5 (Figure ). The appearance of the signal at m/z 409 (Table ) in the pattern of decomposition products of 5 also suggests this route as very probable.
Figure 8. Typical fragmentation for isocyanuric core dendrimers.
It is worth noting that compounds with TRIS moiety decompose in greater number of ways than the other compounds. Moreover, most of the fragmentation patterns described involve decomposition of core structure, which is unlikely for the rest of compounds. This clearly indicates that Tris moiety is less prone to dissociation and other decomposition routes are often preferred. The routes involving common dissociation and/or dehydration (Figure ) are similar to these found for other substances tested.
Figure 9. Common fragmentation pattern of compounds 1 and 4.
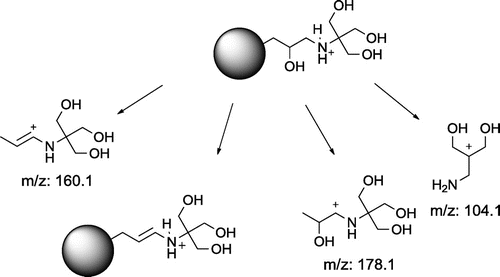
The compounds containing vicinal hydroxyl groups reveal a similar pattern of fragmentation (Figure ), but additionally they can be dehydrated for the second time. Moreover, due to the presence of an tertiary amine, various N-methyleneiminium compounds seem to be formed. Bond cleavage can proceed from both polyol and dendrimer sides, the latter being slightly preferred.
Figure 10. Common fragmentation pattern of compounds 2, 3, 5 and 6 (n=1 or 4).
4. Conclusion
Synthesis of dendrimers G0 using triglycidylic substrates I and II and selected aminopolyols gave expected products with good yield. The molecular masses of obtained products were controlled just after synthesis using MALDI TOF mass spectrometry. The products, whose MALDI mass spectra indicated the presence of side-products were purified by column chromatography. The purified products were characterized by NMR spectra, which confirmed their satisfactory purity and allowed thorough assignment of proton and carbon chemical shift. The samples were further used for MSMS experiment, where [M+H]+ ions were selected as precursors for collision-induced spectra and main fragmentation routes of these ions were investigated.
The group of compounds studied undergoes similar processes of fragmentation and ions formed tend to dissociate at the sites of similar structure, regardless of the structure of the dendrimer core.
A significant variety of core dissociation fragments produced by compounds 1 and 4 suggests that TRIS moiety is not as prone to retain positive charge as N-methylated sugar-like substituents, most probably because of lacking of the N-methyl group. This potentially allows confirmation of attachment of aminopolyols to dendrimers of higher generations. The obtained dendrimers are prospective precursors to the development of larger dendritic structures.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
Support from the National Centre for Research and Development (Poland) under [grant number PBS1/A9/13/2012] is gratefully acknowledged. G. B. and S. J. thank for the support from the National Science Centre (Poland) under research [grant number 2013/11/B/ST3/04190; contract number DEC-2013/11/B/ST3/04190].
References
- Uchegbu IF, Schatzlein AG, editors. Polymers in drug delivery. I editon. Boca Raton, FL: CRC Press; 2006. p. 199–236.10.1201/9781420021677
- Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent SMANCS. Cancer Res. 1986;46:6387–6392. PMID: 2946403.
- Teow HM, Zhou Z, Najlah M, et al. Delivery of paclitaxel across cellular barriers using a dendrimer-based nanocarrier. Int. J. Pharm. 2013;441:701–711. doi:10.1016/j.ijpharm.2012.10.024.
- Najlah M, D’Emanuele A. Crossing cellular barriers using dendrimer nanotechnologies. Curr. Opin. Pharmacol. 2006;6:522–527. doi:10.1016/j.coph.2006.05.004.
- Medina SH, El-Sayed ME. Dendrimers as carriers for delivery of chemotherapeutic agents. Chem. Rev. 2009;109:3141–3157. doi:10.1021/cr900174j.
- Skwarczynski M, Zaman M, Urbani CN, et al. Polyacrylate dendrimer nanoparticles: a self-adjuvanting vaccine delivery system. Angew. Chem. Int. Ed. 2010;49:5742–5745. doi:10.1002/anie.201002221.
- Baigude H, Katsuraya K, Okuyama K, et al. Synthesis of structurally-controlled AIDS vaccine model with glyco-peptide dendrimer scaffolds. Macromol. Chem. Phys. 2004;205:684–691. doi:10.1002/macp.200300097.
- Xu H, Regino CAS, Koyama Y, et al. Preparation and preliminary evaluation of a biotin-targeted, lectin-targeted dendrimer-based probe for dual-modality magnetic resonance and fluorescence imaging. Bioconjugate Chem. 2007;18:1474–1482.10.1021/bc0701085
- Delort E, Darbre T, Reymond J-L. A strong positive dendritic effect in a peptide dendrimer-catalyzed ester hydrolysis reaction. J. Am. Chem. Soc. 2004;126:15642–15643.10.1021/ja044652p
- Yu J, Rajanbabu TV, Parquette JR. Conformationally driven asymmetric induction of a catalytic dendrimer. J. Am. Chem. Soc. 2008;130:7845–7847. doi:10.1021/ja802308a.
- Diallo MS, Arasho W, Johnson JH, et al. Dendritic chelating agents. 2. U(VI) binding to poly(amidoamine) and poly(propyleneimine) dendrimers in aqueous solutions. Environ. Sci. Technol. 2008;42:1572–1579.10.1021/es0715905
- Klementieva O, Benseny-Cases N, Gella A, et al. Dense shell glycodendrimers as potential nontoxic anti-amyloidogenic agents in alzheimer’s disease. Amyloid–dendrimer aggregates morphology and cell toxicity. Biomacromolecules. 2011;12:3903–3909. doi:10.1021/bm2008636.
- Pospisil M, Vannucci L, Fiserova A, et al. Glycodendrimeric ligands of c-type lectin receptors as therapeutic agents in experimental cancer. Adv. Exp. Med. Biol. 2001;495:343–347.10.1007/978-1-4615-0685-0
- Albertazzi L, Serresi M, Albanese A, et al. Dendrimer internalization and intracellular trafficking in living cells. Mol. Pharm. 2010;7:680–688. doi:10.1021/mp9002464.
- Niederhafner P, Sebestik J, Jezek J. Glycopeptide dendrimers. Part I. J. Pept. Sci. 2008;14:2–43.10.1002/(ISSN)1099-1387
- Niederhafner P, Sebestik J, Jezek J. Glycopeptide dendrimers. Part II. J. Pept. Sci. 2008;14:44–65.10.1002/(ISSN)1099-1387
- Niederhafner P, Reinis M, Sebestik J, et al. Glycopeptide dendrimers, part III: a review. Use of glycopeptide dendrimers in immunotherapy and diagnosis of cancer and viral diseases. J. Pept. Sci. 2008;14:556–587. doi:10.1002/psc.1011.
- Aoi K, Itoh K, Okada M. Globular carbohydrate macromolecules “Sugar Balls”. 1. Synthesis of novel sugar-persubstituted poly(amido amine) dendrimers. Macromolecules. 1995;28:5391–5393. doi:10.1021/ma00119a037.
- Gajbhiye V, Palanirajan VK, Tekade RK, et al. Dendrimers as therapeutic agents: a systematic review. J. Pharm. Pharmacol. 2009;61:989–1003. doi:10.1211/jpp/61.08.0002.
- Mishra MK, Kotta K, Hali M, et al. PAMAM dendrimer–azithromycin conjugate nanodevices for the treatment of Chlamydia trachomatis infections. Bioorg. Med. Chem. Lett. 2011;7:935–944. doi:10.1016/j.nano.2011.04.008.
- Schlick KH, Morgan JR, Weiel JJ, et al. Clusters of ligands on dendrimer surfaces. Bioorg. Med. Chem. Lett. 2011;21:5078–5083. doi:10.1016/j.bmcl.2011.03.100.
- Bosnjakovic A, Mishra MK, Ren W, et al. Poly(amidoamine) dendrimer-erythromycin conjugates for drug delivery to macrophages involved in periprosthetic inflammation. Nanomedicine. 2011;7:284–294. doi:10.1016/j.nano.2010.10.008.
- Rico-Lattes I, Blanzat M, Franceschi-Messant S, et al. Catanionic sugar derived surfactants, polymers and dendrimers: from molecules to targeted self-organized systems C. R. Chim. 2005;8:807–814. doi:10.1016/j.crci.2005.02.015.