
ABSTRACT
Structural features such as the shape, the lattice constant, the bond length, the total energy per cell, and the energy bandgap (Eg) of C13H8OS-X are studied by the calculating Partial Density Of States (PDOS), and DOS package of the Material Studio (MS) software. Calculations show that the bond length and the bond angle between atoms insignificant change as 1.316 Å to 1.514 Å for C-C, 1.211 Å for C-O, 1.077 Å to 1.105 Å for C-H; bond angle of round one changes from 118.883° to 121.107° for C-C-C, from 117.199° to 122.635° for H-C-C, from 119.554° to 123.147° for C-C-O and from 109.956° to 117.537° for C-C-H. When C13H8OS-X doped in the order of -Br, -Cu, -Kr, -Ge, -As, and -Fe then bond lengths, bond angles between atoms have a nearly constant value. Particularly for links C-X, there is a huge change in value, respectively 1.876, 1.909, 10.675, 2.025, 2.016, 2.014 Å; the total energy change from Etot = −121,794 eV to Etot = −202,859 eV, and the energy band gap decreases from Eg = 2.001 eV to Eg = 0.915 eV. The obtained results are useful and serve as a basis for future experimental research.
KEYWORDS:
1. Introduction
Polythiophenes are polymer conjugate materials that have been studied and used in many devices such as transistors, opto-electromagnetism, communication equipment, chemical/biological sensors [Citation1–8], light-emitting diodes [Citation9,Citation10], photoelectric cells [Citation11], photoelectron equipment [Citation12,Citation13], water-soluble sensors for detecting DNA, proteins and metal ions [Citation14–16], thermal, optical and biological pigments [Citation17–19] with structural, optical and electrochemical flexibility [Citation20–25].
The reason is that these polymers can assemble spontaneously through intermolecular bonds under the action of a suitable solvent or medium [Citation26–32]. On the other hand, Cui et Kertesz [Citation33] showed the existence of helix polythiophene by the semi-experimental method [Citation33] and other scientific groups showed the existence of polymeric helix [Citation34].
One group of researchers [Citation35–37] has found a polymeric helix structure after the polythiophenes completed bonding with the client molecules and polymers. Other groups of researchers also suggested that non-ionic polythiophenes could fold in hydrophobic solvents [Citation38,Citation39]. These results are determined through Scanning Electron Microscopy (SEM) but they did not observe the diversity of materials [Citation40].
Also, when studying the effect of doped or solvent on the bond lengths of polymer materials, it cannot be studied by X-ray diffraction method or SEM method [Citation41,Citation42].
In recent years, several researchers have used the Density Function Theory (DFT) method to study the structure, electronic structural properties, and transition temperature of conjugated polythiophene derivatives of optical active conjugate polymers [Citation43–47]. Besides that, we have successfully studied the effects of temperature, pressure, atoms number, annealing time on the structure of Al metal [Citation48,Citation49], alloys AlNi [Citation50], NiCu [Citation51], FeNi [Citation52], NiAu [Citation53], polyethylene [Citation54], electronic structure of AuCu [Citation55] and polymers by using DFT method to control band gap by replacing doped -S atoms with -Se atoms [Citation56] or replacing -H atoms with -CH3, -NH2, -NO2 and -Cl [Citation49,Citation57–61] and 4 H-xiclopenta [2,1-b,3; 4-b′] or replacing dithiophene S-oxide with derivatives -O, -S, S = O, -BH2, -SiH2 [Citation47,Citation62–64]. Recently, we have used the DFT method to study the effects of doped groups on the electrical structure and phase transition temperature of monothiophene C13H8OS-X (X = -H, -OH, -Br, -OC2H5, -OCH3). The length of bond C-H = 1.09 Å in C13H8OS-H; the length of bond C-Br = 1.93 Å in C13H8OS-Br; the length of bond C-O = 1.45 Å, the length of bond C-C = 1.51 Å and the length of bond C-H = 1.10 Å in C13H8OS-OC2H5; the length of bond C-O = 1.44 Å and the length of bond O-H = 1.10 Å in C13H8OS-OCH3. The bond angle is 120° for C-C-C, 120° for H-C-C, 120° for C-C-O, 114° for C-S-C, and 109° for S-C-C. Similarly, the bandgap Eg of C13H8OS decreases to Eg = 1.621 eV by doped -Br and increases to 1.646, 1.697, 2.04, and 1.920 eV by replacing with impurities -H, -OH, -OC2H5 or -OCH3. The obtained results show that substituents have a significant influence on the molecular shape, the bond length as well as the frequency range of polythiophene derivatives [Citation57,Citation64].
In this article, we continue to doped atoms of cycle 4 including -H, -Br, -Cu, -Kr, -Ge, -As, and -Fe with the desire to synthesize new polythiophenes with the ability to improve their treatment, environmental stability, and electrical properties.
2. Method of calculation
shows the synthesizing process of poly (C13H8OS-X) (X = -H, -Br, -Cu, -Kr, -Ge, -As, -Fe). The structural and electronic structural properties of poly[3-(3-phenyl prop-1-ene-3-one-1-yl) thiophene] by the calculating Partial Density Of States (PDOS), DOS package of the Material Studio (MS) software, and their transition temperatures were simulated using DFT [Citation65–68] in the framework of DMol3 module [Citation67] in the copyrighted Material Studio software, installed at the Center for Computational Science of the Hanoi National University of Education HNUE (Hanoi, Vietnam). The Generalized Gradient Approximation (GGA) package [Citation69] with the PW91 parametrization for the exchange-correlation function [Citation70,Citation71] and the Monkhorst-Pack [Citation72] k-point sampling were applied into a three-dimensional (3-D) unit cell of poly C13H8OS-H, C13H8OS-Br, C13H8OS-Cu, C13H8OS-Kr, C13H8OS-Ge, C13H8OS-As, C13H8OS-Fe with the lattice constants a = 27 Å, b = 13 Å, c = 6 Å, and the bond angles α = β = γ = 90°. The electron-electrons interaction was described by the Density Function Semi-core Pseudo-Potential [Citation73] and to be considered as a homogeneous electron gas. The tolerance for energy was set at 1 × 10−6 eV, the displacement during the geometry optimization is at level 1 × 10−5 Ha/integer and 5 × 10−3 Å. The synthesis procedure of poly (C13H8OS-X) was shown in .
Figure 1. The synthetic procedure of poly (C13H8OS-X), X is -Br, -Cu, -Fe, -Kr, -As, -Ge
To study the structural features and the bandgap of C13H8OS-X, we use simulations based on the Density Functional Theory (DFT) basis [Citation74,Citation75] included the Schrodinger model [Citation76,Citation77], the Hartree-Fock model [Citation78,Citation79], the Thomas-Fermi model [Citation76], the Hohenberg theorem [Citation76,Citation80,Citation81], and traditional Kohn-Sham theory [Citation76,Citation80,Citation82]. We apply the General Gradient Approximation (GGA) [Citation83], the Korringa-Kohn-Rostocker (KKR) [Citation84], and the Linear-Muffin-Tin-Orbital (LMTO) [Citation85] methods to evaluate calculations.
3. Results and discussion
3.1 Physical properties of C13H8OS-H calculated by molecular dynamics calculations
3.1.1 Structural property of C13H8OS-H
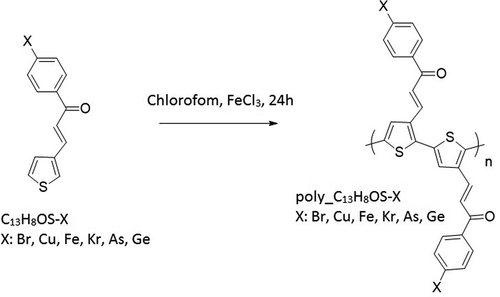
The optimized structures of poly (C13H8OS-H)2 are shown in .
Figure 2. The optimized structures of poly (C13H8OS-H)2 calculated by molecular dynamics calculations
The stable structure of C13H8OS-H after running the NVE (). The final shape of C13H8OS-H has C13H8OS-H poly structures with all C, H, S, O atoms are distributed in a unit cell of the triclinic system with the corresponding cell sizes a = 27.0951 Å, b = 11.5351 Å, c = 6.1176 Å, α = β = 90° and γ = 94.42°. The distance between atoms in round one changes from 1.380 Å to 1.41 Å for C-C, and from 1.060 Å to 1.119 Å for C-H. The distance between round one and round two changes from 1.316 Å to 1.514 Å for C-C, 1.211 Å for C-O, and from 1.077 Å to 1.105 Å for C-H. The obtained results are in good agreement with the structural determination [Citation57,Citation64] for which C-C = 1.33 Å, C-O = 1.23 Å, and previous calculation [Citation57,Citation64]. The bond length in round two changes from 1.372 Å to 1.399 Å for C-C, 1.722 Å for C-S, and from 1.106 Å to 1.148 Å for C-H. The bond angle of round one changes from 118.883° to 121.107° for C-C-C, from 117.199° to 122.635° for H-C-C, from 119.554° to 123.147° for C-C-O, and from 109.956° to 117.537° for C-C-H. The bond angle of round two changes from 111.186° to 115.008° for C-C-C, from 122.321° to 124.347° for H-C-C, 89.695° for C-S-C, from 109.929° to 114.042° for S-C-C, and 122.983° for S-C-H.
3.1.2 Electronic Structure of C13H8OS-H
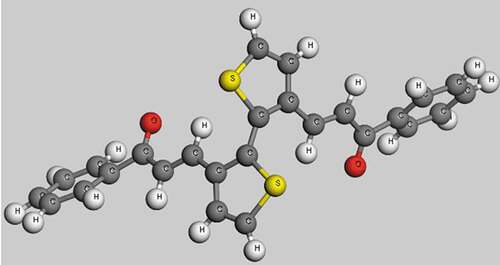
The bandgap is Eg = 2.255 eV (Figure 3a1) and the density of electrons of C13H8OS-H (Figure 3a2) has a maximum value of 23.5%. The density of electrons for C13H8OS-H is shown in .
Table 1. The density of electrons for C13H8OS–H
The results of show that the phase transition temperature zone of C13H8OS-H ranges from 567.5 K to 611.1 K, where the glass temperature Tg = 567.5 K and melting temperature Tm = 611.1 K. These values are larger than values obtained in [Citation57,Citation64]. The reason for this phenomenon is that we put the material into a force field with boundary conditions different from initial boundary conditions. The obtained density of electrons for C13H8OS-H is shown in .
Figure 3. Band structures (a1), Density of states (a2); Phase transition temperature (b) zone for C13H8OS-H
According to , when the energy levels are −20, – 15, – 10, −5, −2.5, 0, 2.5, and 5 eV, the corresponding electron densities of C13H8OS-H are 4.099 %, 3.110 %; 5.631 %, 16.394 %, 5.606 %, 9.703 %, 6.9044 %, and 5.684%.
3.2. The effect of doped on structural features of C13H8OS-X
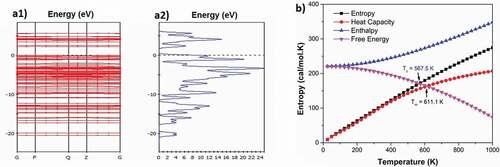
The obtained results show that the distance between the atoms and the bond angle does not change significantly, and that is completely consistent with previous simulations [Citation57,Citation64]. However, the distance between the doped atoms changes strongly for poly(C13H8OS-H) doped. Concretely this distance is 1.116 Å for doped C-H (); 1.876 Å for doped C-Br (); 1.909 Å for doped C-Cu (); 10.675 Å for doped C-Kr (); 2.025 Å for doped C-Ge (); 2.016 Å for doped C-As (); 2.014 Å for doped C-Fe (). Structural features of pure and doped C13H8OS-X, X = H, Br, Cu, Kr, Ge, As, Fe are shown in , respectively.
Table 2. Structural features of pure and doped C13H8OS-X, X = H, Br, Cu, Kr, Ge, As, Fe
Figure 4. Optimized structures of Br-doped (a), Cu-doped (b), Kr-doped (c), Ge-doped (d), As-doped (e) and Fe-doped (f) C13H8OS-X models
3.3. The effect of doped on electronic structures of C13H8OS-X
gives the valence electrons and the value of bandgap energy for doped C13H8OS-X. The bandgap energy for C13H8OS-X doped -H, -Br, -Cu, -Kr, -Ge, -As, -Fe decreases. The values of bandgap energy for doped C13H8OS-X are shown in . The band structure and the density of states for C13H8OS-X doped -H, -Br, -Cu, -Kr, -Ge, -As, -Fe are shown in , describes the density of states for metal-doped C13H8OS-X models with different elements. The obtained results show that the molecules have the shape of box with precise cell sizes a = 27.095 Å, b = 11.535 Å, c = 6.118 Å for C13H8OS-H; a = 25.305 Å, b = 12.398 Å, c = 6.070 Å for C13H8OS-Br; a = 25.678 Å, b = 13.049 Å, c = 5.928 Å for C13H8OS-Cu a = 26.092 Å, b = 12.970 Å, c = 6.064 Å C13H8OS-Kr; a = 26.160 Å, b = 12.799 Å, c = 6.146 Å for C13H8OS-Ge; a = 26.348 Å, b = 12.913 Å, c = 6.112 Å for C13H8OS-As and a = 24.400 Å, b = 13.662 Å, c = 5.993 Å for C13H8OS-Fe. The bond angles of different poly(C13H8OS-X) derivatives are 123.017° with C-C-H of C13H8OS-H; 115.798° with C-C-Br of C13H8OS-Br; 120.957° with C-C-Cu of C13H8OS-Cu; 123.017° with C-C-Br of C13H8OS-Kr; 120.617° with C-C-Ge of C13H8OS-Ge; 125.009° with C-C-As of C13H8OS-As and 11.022° with C-C-Fe of C13H8OS-Fe. When the energy levels are −20, −15, −10, −7.5, −5, 0, 5 and 7.5 eV, the corresponding densities of electrons for C13H8OS-H are 4.099 %, 3.110 %, 5.631 %, 16.394 %, 5.606 %, 9.703 %, 6.044 % and 5.684 %.
Table 3. Bandgap values of pure and doped C13H8OS-X
Figure 5. Band structure and density of states of Br-doped (a), Cu-doped (b), Kr-doped (c), Ge-doped (d), As-doped (e), and Fe-doped (f) with C13H8OS-X
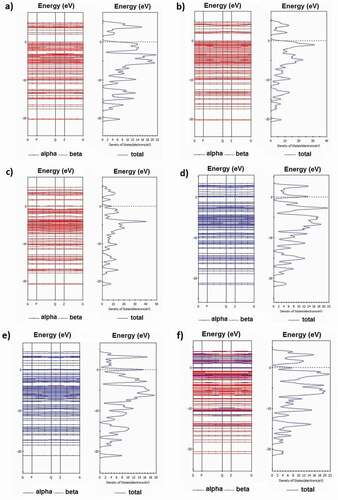
Figure 6. Bandgap values of metal-doped C13H8OS-X models with different elements
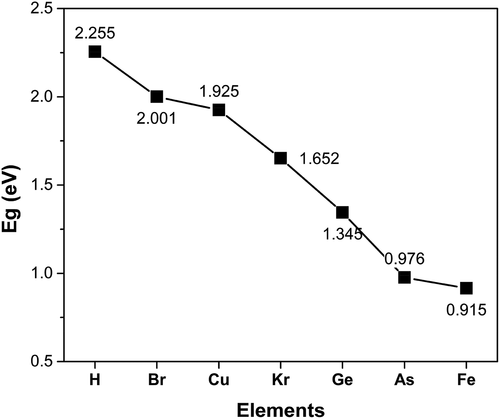
Figure 7. The density of states of metal-doped C13H8OS-X models with different elements
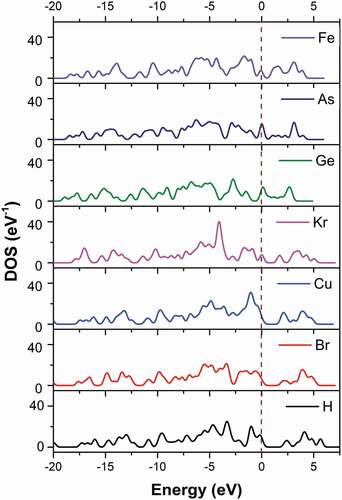
The density of electrons for C13H8OS-X doped the functional groups -Br, -Cu, -Kr, -Ge, -As, -Fe changes greatly. For example when the energy level at −20 eV, the density of electrons is 4.099% for C13H8OS-X doped -H, 3.262% for C13H8OS-X doped -Br,4.071% for C13H8OS-X doped -Cu, 0,014% for C13H8OS-X doped -Kr, 0% for C13H8OS-X doped -Ge and -As, 0.063% for C13H8OS-X doped – Fe. At −15 eV, the corresponding densities of electrons are 3.110%, 10.772%, 6.619%, 2.511%, 11.541%, 9.02% and 3.785%. At −10 eV, the corresponding densities of electrons are 5.631%, 11.681%, 8.704%, 10.836%, 7.388%, 3.312% and 7.529%. At-5 eV, the corresponding densities of electrons are 16.394%, 18.743%, 21.630%, 17.866%, 17.608%, 17.583% and 15.650%; At −2.5 eV, the corresponding densities of electrons are 5.606%, 3.954%, 9.327%, 3.585%, 15.764%, 12.443% and 11.425%. At 0, the corresponding densities of electrons are 9.703%, 6.343%, 7.909%, 7.806%, 12.238%, 15.230% and 7.020%. At 2.5 eV, the corresponding densities of electrons are 6.044%, 3.857%, 5.089%, 6.786%, 12.214%, 2.208% and 6.016%. At 5 eV, the corresponding densities of electrons are 5.684%, 6.493%, 3.290%, 3.903%, 0, 0 and 0. This shows that in the valence region, the density of electrons has the largest percentage extending the maximum value at the energy range of −5 eV. Also, the bandgap Eg decreases for the doped in the order of -Br, -Cu, -Kr, -Ge, -As, and -Fe. Besides, the total energy Etot of the system decreases suddenly at -Kr, which shows that the Etot of the −4p subclass of the material decreases for increasing the subclass from −4p2 to −4p3, −4p5, and −4p6. The obtained results are used as the basis for future experimental research and used to predict the structural features and electronic structure of C13H8OS-X doped.

4. Conclusion
The effects of doped -H, -Br, -Cu, -Kr, -Ge, -As, and -Fe on structural features and the electronic structure of C13H8OS-X are studied by the GGA package PW91 of the Material Studio software copyrighted based on the DFT method. Structural features such as the lengths of the bonds C-H, C-Fe, C-Ge, C-C, C-Br, C-O, C-Cu, C-Kr, C-As and the bond angles H-C-H, H-C-C, C-C-C, C-Fe-H, C-Ge-C, C-Br-C, C-O-C, Cu-C-C, Kr-C-C, As-C-C do not change significantly. However, when then total energy change of system decrease from Etot = −121,794 eV to Etot = −202,859 eV; and the total energy and the bandgap of C13H8OS-X doped in the order of -H, -Br, -Cu, -Kr, -Ge, -As and -Fe decrease from Etot = – 121,794 eV to Etot = – 202,859 eV and from Eg = 2.001 eV to Eg = 0.915 eV respectively. This shows that the influence of benzene ring and impurities on electronic structure features and the bandgap of poly materials is important and these results are considered as a basis for future experimental research.
Disclosure statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper. The funders had no role in the design of the study and the authors declare no conflict of interest.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Additional information
Funding
References
- Brown DM, Zeef LAH, Ellis J, et al. Identification of novel genes in Arabidopsis involved in secondary cell wall formation using expression profiling and reverse genetics.. The Plant Cell. 2005;17(8):2281–2305.
- Ho H-A, Najari A, Leclerc M. Optical detection of DNA and proteins with cationic polythiophenes. Acc Chem Res. 2008;41(2):168–178.
- Nielsen CB, McCulloch I. Recent advances in transistor performance of polythiophenes. Prog Polym Sci. 2013;38(12):2053–2069.
- Agbolaghi S, Zenoozi S. A comprehensive review on poly(3-alkylthiophene)-based crystalline structures, protocols and electronic applications. Organic Electron. 2017;51:362–403.
- Pathiranage DSD, Taniya MSK, Niermann CN, et al. Role of polythiophenes as electroactive materials Part A Polymer chemistry. J Polym Sci. 2017;55(20):3327–3346.
- Ibanez JG, Rincón ME, Gutierrez-Granados S, et al. Conducting polymers in the fields of energy, environmental remediation, and chemical–chiral sensors. Chem Rev. 2018;118(9):4731–4816.
- Lévesque I, Bazinet P, Roovers J. Optical properties and dual electrical and ionic conductivity in poly(3-methylhexa(oxyethylene)oxy-4-methylthiophene). Macromolecules. 2000;33(8):2952–2957.
- Snook GA, Kao P, Best AS. Conducting-polymer-based supercapacitor devices and electrodes. J Power Sources. 2011;196(1):1–12.
- Touhami G, Benattilah A, Youcef S, et al. Realization and characterization of p-typed polythiophene based organic photovoltaic cells, journal of nano- and electronic physics. 2018;10(1):01008(5)
- Maity N, Ghosh R, Nandi AK. Optoelectronic properties of self-assembled nanostructures of polymer functionalized polythiophene and graphene. Langmuir. 2018;34(26):7585–7597.
- Borrelli DC, Lee S, Gleason KK. Optoelectronic properties of polythiophene thin films and organic TFTs fabricated by oxidative chemical vapor deposition. J Mater Chem C. 2014;2(35):7223–7231.
- Wang F, Li M, Wang B, et al. Synthesis and characterization of water-soluble polythiophene derivatives for cell imaging. Sci Rep. 2015;5(1):7617.
- Vázquez-Arce A, Zaragoza-Galán G, Aguilar-Ortíz E, et al. Luminescent polythiophenes-containing porphyrin units: synthesis, characterization, and optical properties. Des Monomers Polym. 2014;17(1):78–88.
- Sadeghi K, Yoon J-Y, Seo J. Chromogenic polymers and their packaging applications: a review. Polymer Rev. 2020;60(3):442–492.
- Elbing M, Garcia A, Urban S, et al. In situ conjugated polyelectrolyte formation. Macromolecules. 2008;41(23):9146–9155.
- Aiello S, Wells G, Stone EL, et al. Synthesis and biological properties of benzothiazole, benzoxazole, and chromen-4-one analogues of the potent antitumor agent 2-(3,4-dimethoxyphenyl)-5-fluorobenzothiazole (PMX 610, NSC 721648). J Med Chem. 2008;51(16):5135–5139.
- Cho Y, Ioerger TR, Sacchettini JC. Discovery of novel nitrobenzothiazole inhibitors for Mycobacterium tuberculosis ATP phosphoribosyl transferase (HisG) through virtual screening. J Med Chem. 2008;51(19):5984–5992.
- Radhakrishnan S, Parthasarathi R, Subramanian V, et al. Structure and properties of polythiophene containing hetero aromatic side chains. Comput Mater Sci. 2006;37(3):318–322.
- Radhakrishnan S, Somanathan N. Poly(thiophenes) functionalised with thiazole heterocycles as electroluminescent polymers. J Mater Chem. 2006;16(29):2990–3000.
- Ikai T, Takayama K, Wada Y, et al. Synthesis of a one-handed helical polythiophene: a new approach using an axially chiral bithiophene with a fixed syn-conformation. Chem Sci. 2019;10(18):4890–4895.
- Valderrama-García BX, Rodríguez-Alba E, Morales-Espinoza EG, et al. Synthesis and characterization of novel polythiophenes containing pyrene chromophores: thermal, optical and electrochemical properties. Molecules. 2016; 21(2):172(18).
- Pina J, Rodrigues ACB, Alnady M, et al. Restricted aggregate formationontetraphenylethene-substituted polythiophenes.J Phys Chem C. 2020;124(25):13956–13965.
- Gupta S, Chatterjee S, Zolnierczuk P, et al. Impact of local stiffness on entropy driven microscopic dynamics of polythiophene. Sci Rep. 2020;10(1):9966.
- Kaloni TP, Giesbrecht PK, Schreckenbach G, et al. Polythiophene: from fundamental perspectives to applications. Chem Mater. 2017;29(24):10248–10283.
- Hu Z, Zhang S, Zhang C, et al. Donor–acceptor units modulate the electronic and photoluminescence characteristics of thiophene oligomers. J Appl Phys. 2019;126(24):245501.
- Langeveld-Voss BMW, Janssen RAJ, Meijer EW. On the origin of optical activity in polythiophenes☆in honour of professor Giuseppe Zerbi on the occasion of his 65th birthday. J Mol Struct. 2000;521(1):285–301.
- Hoeben FJ, Jonkheijm P, Meijer EW, et al. About supramolecular assemblies of pi-conjugated systems. Chem Rev. 2005;105(4):1491–1546.
- Kane-Maguire LAP, Wallace GG. Chiral conducting polymers. Chem Soc Rev. 2010;39(7):2545–2576.
- Verswyvel M, Koeckelberghs G. Chirality in conjugated polymers: when two components meet. Polym Chem. 2012;3(12):3203–3216.
- Matsushita S, Jeong YS, Akagi K. Electrochromism-driven linearly and circularly polarised dichroism of poly(3,4-ethylenedioxythiophene) derivatives with chirality and liquid crystallinity. Chem Comm. 2013;49(19):1883–1890.
- Yashima E, Ousaka N, Taura D, et al. Supramolecular helical systems: helical assemblies of small molecules, foldamers, and polymers with chiral amplification and their functions. Chem Rev. 2016;116(22):13752–13990.
- Wang P, Jeon I, Lin Z, et al. Insights into magneto-optics of helical conjugated polymers. J Am Chem Soc. 2018;140(20):6501–6508.
- Cui CX, Kertesz M. Two helical conformations of polythiophene, polypyrrole, and their derivatives. Phys Rev B Condens Matter. 1989;40(14):9661–9670.
- Nilsson KPR, Olsson JDM, Konradsson P, et al. Enantiomeric substituents determine the chirality of luminescent conjugated polythiophenes. Macromolecules. 2004;37(17):6316–6321.
- Haraguchi S, Numata M, Li C, et al. Circularly polarized luminescence from supramolecular chiral complexes of achiral conjugated polymers and a neutral polysaccharide. Chem Lett. 2009;38(3):254–255.
- Shiraki T, Dawn A, Tsuchiya Y, et al. Thermo- and solvent-responsive polymer complex created from supramolecular complexation between a helix-forming polysaccharide and a cationic polythiophene. J Am Chem Soc. 2010;132(39):13928–13935.
- Tamaru S-I, Hori K, Shinkai S. Environment-induced Sequential Interconversion of Amphoteric β-1,3-Glucans/Polythiophene Complexes: a Unique System Applicable to a Naked-eye Detectable Fluorogenic pH Probe. Chem Lett. 2015;44(12):1667–1669.
- Matthews JR, Goldoni F, Schenning APHJ, et al. Non-ionic polythiophenes: a non-aggregating folded structure in water. Chem Comm. 2005;44(1): 5503-5505.
- Leysen P, Teyssandier J, De Feyter S, et al. Controlled Synthesis of a Helical Conjugated Polythiophene. Macromolecules. 2018;51(9):3504–3514.
- Zhang HH, Ma CX, Bonnesen PV, et al. Helical Poly(5-alkyl-2,3-thiophene)s: controlled Synthesis and Structure Characterization. Macromolecules. 2016;49(13):4691–4698.
- Kaspar P, Sobola D, Částková K, et al. Characterization of Polyvinylidene fluoride (PVDF) electrospun fibers doped by carbon flakes. Polymers (Basel). 2020;12:1–15.
- Castkova K, Kastyl J, Sobola D, et al. Structure-Properties relationship of electrospun PVDF fibers. Nanomaterials (Basel). 2020;10(6):1–19.
- Ikai T, Minami S, Awata S, et al. Helicity control of π-conjugated foldamers containing d-glucose-based single enantiomeric units as a chiral source. Polym Chem. 2018;9(46):5504–5510.
- Shokuhi Rad A, Esfahanian M, Ganjian E, et al. The polythiophene molecular segment as a sensor model for H2O, HCN, NH3, SO3, and H2S: a density functional theory study. J Mol Model. 2016;22(6):127.
- Saha A, Ganguly B. A DFT study to probe homo-conjugated norbornylogous bridged spacers in dye-sensitized solar cells: an approach to suppressing agglomeration of dye molecules. RSC Adv. 2020;10(26):15307–15319.
- Ansari MA, Mohiuddin S, Kandemirli F, et al. Synthesis and characterization of poly(3-hexylthiophene): improvement of regioregularity and energy band gap. RSC Adv. 2018;8(15):8319–8328.
- Arjunan V, Thirunarayanan S, Durga Devi G, et al. Substituent influence on the structural, vibrational and electronic properties of 2,5-dihydrothiophene-1,1-dioxide by experimental and DFT methods. Spectrochim Acta A Mol Biomol Spectrosc. 2015;150:641–651.
- Nguyen-Trong D, Nguyen-Tri P. Understanding the heterogeneous kinetics of Al nanoparticles by simulations method. J Mol Struct. 2020;1218:128498.
- Quoc TT, Trong DN. Molecular dynamics factors affecting on the structure, phase transition of Al bulk. Phys B Condens Matter. 2019;570:116–121.
- Nguyen-Trong D, Nguyen-Tri P. Factors affecting the structure, phase transition and crystallization process of AlNi nanoparticles. J Alloys Compd. 2020;812:152133.
- Nguyen-Trong D, Nguyen-Tri P. Molecular dynamic study on factors influencing the structure, phase transition and crystallization process of NiCu6912 nanoparticle. Mater Chem Phys. 2020;250:123075.
- Nguyen-Trong D, Pham-Huu K, Nguyen-Tri P. Simulation on the factors affecting the crystallization process of FeNi alloy by molecular dynamics. ACS Omega. 2019;4(11):14605–14612.
- Trong DN, Long VC, Ţălu S. The structure and crystallizing process of NiAu alloy: a molecular dynamics simulation method. J Composites Sci. 2021;5(1):1–14.
- Trong DN, Quoc TT, Minh HDT, et al. Structure, plastic deformation of polyethylene: a molecular dynamics method. Adv Mater Phys Chem. 2020;10:125–150.
- Nguyen-Trong D, Nguyen-Chinh C, Duong-Quoc V. Study on the Effect of Doping on Lattice Constant and Electronic Structure of Bulk AuCu by the Density Functional Theory. Journal of Multiscale Modelling, 2020;11(02):2030001.
- Rittmeyer SP, Groß A. Structural and electronic properties of oligo- and polythiophenes modified by substituents. Beilstein J Nanotechnol. 2012;3:909–919.
- Vu Quoc T, Thi Thuy DT, Thanh TD, et al. Some chalcones derived from thio-phene-3-carbaldehyde: synthesis and crystal structures, acta crystallographica. section e, crystallographic communications. 2019;75(Pt7):957–963.
- Vu QT, Pavlik M, Hebestreit N, et al. Nanocomposites based on titanium dioxide and polythiophene: structure and properties. React Funct Polym. 2005;65(1–2):69–77.
- Vu Q-T, Pavlik M, Hebestreit N, et al. Electrophoretic deposition of nanocomposites formed from polythiophene and metal oxides. Electrochim Acta. 2005;51(6):1117–1124.
- Trung VQ, Linh NN, Linh DK, et al. Synthesis and characterization of polythiophenes from hydrazone derivatives sidegroups. Vietnam Journal of Chemistry, 2016;54(6):730-735.
- Banjo S, Ayobami OO, Ajibade AI. Structural and electronic properties of 4H-cyclopenta[2,1-b,3;4-b’]dithiophene S-oxide (BTO) derivatives with an S, S=O, O, SiH2, or BH2 bridge: semi-empirical and DFT study. J Mol Model. 2012;18(6):2755–2760.
- Lo P-K, Lau K-C. High-level ab initio predictions for the ionization energies and heats of formation of five-membered-ring molecules: thiophene, Furan, Pyrrole, 1,3-Cyclopentadiene, and Borole, C4H4X/C4H4X+ (X = S, O, NH, CH2</sub>, and BH). J Phys Chem A. 2011;115(5):932–939.
- Barlow S, Odom SA, Lancaster K, et al. Electronic and optical properties of 4H-Cyclopenta[2,1-b:3,4-b′]bithiophene derivatives and Their 4-heteroatom-substituted analogues: a joint theoretical and experimental comparison. J Phys Chem A. 2010;114(45):14397–14407.
- Vu QT, Tran TT, Nguyen TC, et al. DFT prediction of factors affecting the structural characteristics, the transition temperature and the electronic density of some new conjugated polymers. Polymers (Basel). 2020;12(6):1207(13).
- Pettifor DG, Cottrell AH. Electron theory in alloy design. Leed, UK: Maney Publishing; 1992.
- Kresse G, Hafner J. Ab initio molecular dynamics for liquid metals. Phys Rev B. 1993;47(1):558–561.
- Delley B. An all‐electron numerical method for solving the local density functional for polyatomic molecules. J Chem Phys. 1990;92(1):508–517.
- Bernholc J. Computational materials science: the era of applied quantum mechanics. Physics Today 1999;52(9):30-35.
- Perdew JP, Burke K, Ernzerhof M. Generalized gradient approximation made simple. Phys Rev Lett. 1996;77:3865–3868.
- Tsuzuki S, Lüthi HP. Interaction energies of van der Waals and hydrogen bonded systems calculated using density functional theory: assessing the PW91 model. J Chem Phys. 2001;114(9):3949–3957.
- Perdew JP, Wang Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys Rev B. 1992;45(23):13244–13249.
- Monkhorst HJ, Pack JD. Special points for Brillouin-zone integrations. Phys Rev B. 1976;13:5188–5192.
- Hamann DR, Schlüter M, Chiang C. Norm-conserving pseudopotentials. Phys Rev Lett. 1979;43:1494–1497.
- Itoh A, Takahashi Y, Furukawa T, et al. Solid-state calculations of poly(vinylidene fluoride) using the hybrid DFT method: spontaneous polarization of polymorphs. Polym J. 2014;46(4):207–211.
- Yoshida Y, Mawatari Y, Tabata M. Proposed mechanism for the high-yield polymerization of oxyethyl propiolates with rh complex catalyst using the density functional theory method. Polymers. 2019;11(1):93.
- Parr RG, Yang W. Density-functional theory of atoms and molecules. New York: Oxford University Press; 1989.
- Koch W, Holthausen MC. A Chemist’s Guide to Density Functional Theory. 2nd ed. Wiley; wiley-vch, 2001.
- Søndenå R, Stølen S, Ravindran P, et al. Corner- versus face-sharing octahedra in AMnO3 perovskites (A=Ca, Sr, and Ba). Phys Rev B. 2007;75(18):184105.
- Slater JC. A simplification of the hartree-fock method. Phys Rev. 1951;81(3):385–390.
- Hohenberg P, Kohn W. Inhomogeneous electron gas. Phys Rev. 1964;136(3B):B864–B871.
- Englisch H, Englisch R. Hohenberg-Kohn theorem and non-V-representable densities. Phys A Stat Mech Appli. 1983;121(1):253–268.
- Kohn W, Sham LJ. Self-consistent equations including exchange and correlation effects. Phys Rev. 1965;140(4A):A1133–A1138.
- Wang Y, Perdew JP. Correlation hole of the spin-polarized electron gas, with exact small-wave-vector and high-density scaling. Phys Rev B. 1991;44(24):13298–13307.
- Ruban AV, Skriver HL, Nørskov JK. Surface segregation energies in transition-metal alloys. Phys Rev B. 1999;59(24):15990–16000.
- Andersen OK, Jepsen O, Glotzel D, Canonical description of the band structures of metals, in: Bassani FF, Tosi MP (Ed.) Proceedings of the International School of Physics ‘Enrico Fermi’, Varenna, Italy, 1985;1(7000): 1985(59-176).