
ABSTRACT
The content of this paper focuses/shed light on the effects of X (X = S in P1 and X = O in P2) in C11H7NSX and R (R = H in P3, R = OCH3 in P4, and R = Cl in P5) in C18H9ON2S2-R on structural features and band gaps of the polythiophenes containing benzo[d]thiazole and benzo[d]oxazole by the Density Function Theory (DFT) method/calculation. The structural features including the electronic structure lattice constant (a), shape, total energy (Etot) per cell, and link length (r), are measured via band gap (Eg) prediction with the package of country density (PDOS) and total country density (DOS) of material studio software. The results obtained showed that the link angle and the link length between atoms were not changed significantly while the Etot was decreased from Etot = – 1904 eV (in P1) to Etot = – 2548 eV (in P2) when replacing O with S; and the Etot of P3 was decreased from Etot = – 3348 eV (in P3) when replacing OCH3, Cl on H of P3 corresponding to Etot = – 3575 eV (P4), – 4264 eV (P5). Similarly, when replacing O in P1 with – S to form P2, the Eg of P1 was dropped from Eg = 0.621 eV to Eg = 0.239 eV for P2. The Eg of P3, P4, and P5 is Eg = 0.006 eV, 0.064 eV, and 0.0645 eV, respectively. When a benzo[d]thiazole was added in P1 (changing into P3), the Eg was extremely strongly decreased, nearly 100 times (from Eg = 0.621 eV to Eg = 0.006 eV). The obtained results serve as a basis for future experimental work and used to fabricate smart electronic device.
Graphical abstract
1. Introduction
In recent years, polythiophene-containing heterocycles have many advanced applications based on their high environmental sustainability, structural flexibility, optical stability, and electrochemical characteristics [Citation1–12]. They were reported as potential functional materials, such as organic field-effect transistors [Citation13,Citation14], organic light-emitting diodes [Citation15,Citation16], organic photovoltaic cells [Citation17], and other optoelectronic devices [Citation18,Citation19]. Moreover, they also have many applications in pharmacology as water-soluble sensing agents for the recognition of DNA, proteins, and metal ions [Citation20–22], thermochromism, photochromism, and biochemist [Citation23–25]. Among those, benzothiazole-based polythiophenes have attracted much attention thanks to their wide range of biological activities [Citation26,Citation27]. A novel conducting poly[3-(benzothiazole-2-yl)] thiophene polymerized by electrochemical and chemical synthesis has been studied for its optical absorption and photoluminescence characteristics [Citation28–30]. Some technologically advanced methods were applied for the synthesis of 3-(benzothiazole-2-yl)thiophene from the reaction of thiophene-3-carbaldehyde with o-amino thiophenol in refluxing ethanol [Citation31] or under microwave radiation without solvent and catalyst [Citation32]. However, very few studies based on the molecular orbital calculations have been performed for oligothiophenes containing heteroaromatic side chains. Basing on theoretical prediction, Radhakrishnan S. et al. using suggested the structure optical properties relationship of oligothiophenes having four thiophene units [Citation27,Citation33]. In general, the periodic calculations of geometrical stability and electrical properties of the polythiophenes are often unattended with their published experimental data while these theoretical properties are important bases for deeply understanding the nature and application ability in the energy industry and electrically conducting materials. Therefore, structural analysis based on the theoretical calculation of polythiophene derivatives is very necessary. Among theoretical quantum mechanical methods, Density Functional Theory (DFT) method is well-known as an effective method for evaluation of the transition temperature, electronic properties, and structural characteristics of the π-conjugated polythiophene derivatives [Citation34–42]. However, only a few studies on the electronic structures of polymers using the DFT method have been done to control the band gap for determining the alternation between conductors and insulators in solar cells, diodes, or transistors. A popular pathway to synthesize new polythiophene derivatives is substitution in polythiophenes, for example, replacement of S atom with Se or Te atoms [Citation43] or replacement of H atoms with CH3, NH2, NO2, or Cl [Citation44]. The DFT method has been applied for the assessment of structural and electronic characteristics of 4 H-cyclopenta[2,1-b,3;4-b′] dithiophene S-oxide derivatives including X (X: O, S, S = O, BH2, SiH2) as a bridge [Citation45]. The other five-membered ring molecules and ionization energies (IEs) and the heats of formation of thiophene were calculated and reached a high precision level of ab initio predictions [Citation46]. Recently, the team members also studied the factors affecting the structural, mechanical, and magnetic properties of metal Fe [Citation47,Citation48], Al [Citation49–51], Ni [Citation52–54], Ag [Citation55], alloys AlNi [Citation56], NiCu [Citation57,Citation58], FeNi [Citation59,Citation60], AgAu [Citation61], NiAu [Citation62], and replace the H derivatives of poly C13H8OS-H with metal atoms Br, Cu, Kr, Ge, As, Fe [Citation63] showed that the Eg band gap decreased, leading to an increase in the conductivity. The obtained results will contribute to research to find materials new for application in the industrial age. In addition, the electronic and optical characteristics of 4 H-cyclopenta[2,1-b:3,4-b′]bithiophene derivatives combining with a variety of functional groups including carbon atoms and heteroatoms in the 4-position were predicted using DFT calculations [Citation64]. Along with that, the energy band gap (Eg) of C13H8OS was decreased to Eg = 1.621 eV when doping with Br, while the energy band gaps of C13H8OS was increased to Eg = 1.646, 1.697, 2.04, and 1.920 eV, respectively, when doping with H, OH, OC2H5, or OCH3 groups. The obtained results proved that the substituents had a remarkable effect on the link length as well as band gap of polythiophene derivatives, and molecular shape. More recently, our research group studied some novel polythiophenes containing benzo[d]thiazole that presented a catalyst- and solvent-free microwave-assisted synthesis of mono thiophene [Citation8]. Their structure and properties were determined by FT-IR, 1H-NMR, 13C-NMR spectra, single-crystal X-ray diffraction, and the TGA method [Citation32]. We have also mentioned the chemical polymerization of the monomers using anhydrous FeCl3 as an oxidant in anhydrous chloroform. However, the structures, phase transition temperature, and electronic property data of these polymers are rather limited. The purpose of this work is to predict the theoretical structure and properties in the ideal state of some newly synthesizing polythiophenes containing benzo[d]thiazole and benzo[d]oxazole using DFT calculation. With the available results, we are looking forward to synthesize these polythiophenes using chemical polymerization to environmental stability, improve their processability, and electrical properties.
2. Computational methods
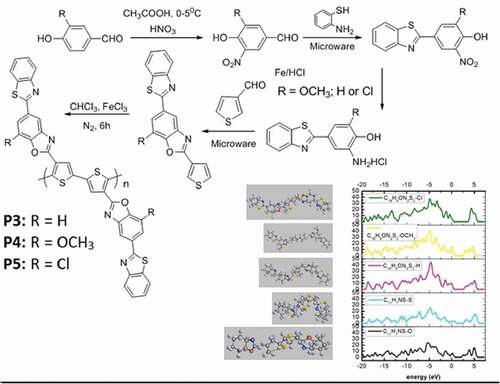
The five polythiophenes containing benzo[d] thiazole and benzo[d]oxazole have been formulated and synthesized according to the process of Material Studio software, such as: Polymer 1 is poly[3-(benzo[d]thiazole-2-yl)thiophene], P1: C11H7NS-O; Polymer 2 is poly [3-(benzo[d] oxazole-2-yl)thiophene], P2: C11H7NS-S; Polymer 3 is poly-3-(5-(benzo[d] thiazole-2-yl)-7-methoxybenzo[d]oxazole-2-yl)thiophene, P3: C18H9ON2S2-H; Polymer 4 is poly-3-(5-(benzo[d]thiazole-2-yl)benzo[d]oxazole-2-yl)thiophene, P4: C18H9ON2S2-OCH3; Polymer 5 is poly-3-(5-(benzo[d]thiazole-2-yl)-7-chlorobenzo[d] oxazole-2-yl)thiophene), P5: C18H9ON2S2-Cl ().
Figure 1. The synthetic procedure of the five novel polythiophenes containing benzo[d]thiazole and benzo[d]oxazole
![Figure 1. The synthetic procedure of the five novel polythiophenes containing benzo[d]thiazole and benzo[d]oxazole](/cms/asset/46468ae6-190e-485b-9c82-0e757ca24d11/tdmp_a_1971376_f0001_b.gif)
To study the band gap, the structural features of the polymers, we have used the DMol3 in the copyrighted Material Studio software combined with the Density Function Theory (DFT) method. To investigate band gap, the structural, calculates the thermodynamic properties of many different polymer systems [Citation65–71], and polythiophenes [Citation72,Citation73]. Besides, use the GGA package [Citation74] with takes the combined parameters of the PW91 exchange, correlation functional [Citation75,Citation76], and the K-point grid according to the Monkhorst–Pack diagram [Citation77] was directed into a three-dimensional unit cell with definite dimensions a, b, c, α, β and γ as follows: P1: C11H7NS-O (a = 10 Å, b = 10 Å, c = 25 Å), P2: C11H7NS-S (a = 10 Å, b = 10 Å, c = 6 Å), P3: C18H9ON2S2-H (a = 10 Å, b = 10 Å, c = 40 Å), P4: C18H9ON2S2-OCH3 (a = 10 Å, b = 15 Å, c = 40 Å) and P5: C18H9ON2S2-Cl (a = 10 Å, b = 15 Å, c = 40 Å) and α = β = γ = 90°. The electrons are regarded in a homogeneous state in a system of interacting electrons through the Density Function semi-core pseudopotential [Citation60]. During optimization of the displacement, the geometry at level 1 × 10−5 Ha/integer and 5 × 10−3 Å; and the energy was established at 1 × 10−6 eV. The whole process of calculating and analyzing data was based on copyrighted Material Studio software, which was set up at the Center for Computational Science of Hanoi University of Education, Vietnam.
3. Results and discussion
3.1. Effect of the material
The structural features and band gap of poly C11H7NS-O (P1), C18H9ON2S2-H (P3) are shown in and .
Figure 2. Structured shape, electronic structure of P1: C11H7NS-O (a1, b1), P3: C18H9ON2S2-H (a2, b2)
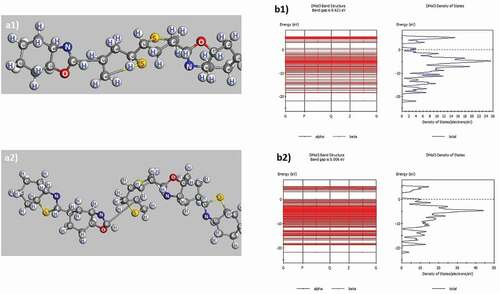
Table 1. Structural characteristic quantities of P1 and P3
The obtained results show that in C11H7NS-O (P1) molecule, the length of C–H links is between 1.106 Å ÷ 1.108 Å, C–N is 1.459 Å ÷ 1.430 Å, C–S is 1.883 Å ÷ 1.924 Å, C–C is 1.532 Å ÷ 1.545 Å, and the link angle H–C–H is in the range of 106.260° and 106.052°, H–C–C is 109.169° ÷ 109.209°, C–C–C is 111.48° ÷ 111.457°, C–N–H is 107.658° ÷ 109.344°, C–S–C is 105.860° ÷ 105.902°, C–N–C is 86.808°, C–O–C is 107.237°. Similarly, in the C18H9ON2S2-H (P3) molecule, the C–H link length is 1.104 Å ÷ 1.105 Å, C–N is 1.419 Å ÷ 1.435 Å, C–S is 1.895 Å ÷ 1.991 Å, C–C is 1.520 Å ÷ 1.546 Å, C–O is 1.459 Å ÷ 1.483 Å, and link angle H–C–H is 105.910° ÷ 106.646°, H–C–C is 110.042° ÷ 110.867°, C–C–C is 108.997° ÷ 111.613°, H–C–N is 108.381° ÷ 109.471°, C–S–C is 86.276° ÷ 91.577°, C–N–C is 111.630° ÷ 112.273°. The results obtained on the link lengths are completely consistent with the previously published results [Citation47]. Besides, the total energy of the system (Etot) of the P1 and P3 is Etot = – 1904 eV and Etot = – 3348 eV, respectively. When adding benzo[d]thiazole to the P1, the link angle and the link length of the P3 between the atoms as well as the benzene ring did not change significantly. This indicated that the structure of P1 did not change when it was added to a benzene ring although benzene ring can lead to a change in the shape, size, and total energy of the Etot system (a1, a2) corresponding to base cell size with wide-band gap decreased from Eg = 0.6210 eV to Eg = 0.0060 eV (). These results are important bases for predicting the structure of polythiophenes containing benzo[d]thiazole and benzo[d]oxazole as well as the change of their Etot and Eg.
3.2. Effects of doping
The P1 and P2 polymers were chosen as the basic materials, then their composition is changed as follows: O was substituted with S in P1: C11H7NS-O to get P2: C11H7NS-S. Similarly, replacing H with OCH3 in P3: C18H9ON2S2-H to get P4: C18H9ON2S2-OCH3 and Cl to get P5: C18H9ON2S2-Cl was done. The molecular geometry and band gap of the above samples are displayed in and .
Figure 3. Structured shape, electronic structure of P1: C11H7NS-O (a1, b1), P2: C11H7NS-S (a2, b2), P3: C18H9ON2S2-H (a3, b3), P4: C18H9ON2S2-OCH3 (a4, b4), P5: C18H9ON2S2-Cl (a5, b5)
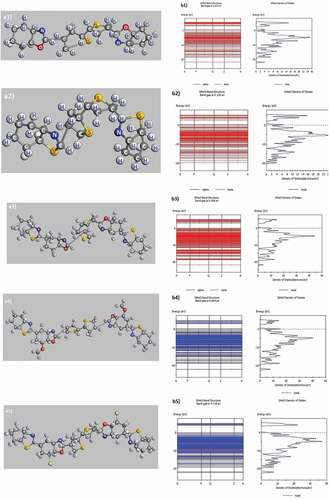
Table 2. Structural characteristics of polymers (P1-P5) with different impurities
The network constant values such as a, b, c, α, β, and γ of the P1-P5 samples in equilibrium show that their network constant values and link angle are not changed significantly. The structural features of P1 and P2 have been reported in a1, b1, a3, b3, . Similarly, for the P2: C11H7NS-S has link lengths in the following range: C–H of 1.14 Å, C–N of 1.412 Å ÷ 1.448 Å, C–S of 1.880 Å ÷ 1.994 Å, C–C of 1.536 Å ÷ 1.541 Å and the bond angle between the atoms as H–C–H of 96.916° ÷ 110.185°, H–C–C of 109.135° ÷ 109.471°, C–C–C of 109.135° ÷ 113.254°, C–N–H of 108.381° ÷ 109.471°, C–S–C of 86.276° ÷ 91.577°, C–N–C of 111.630° ÷ 112.273° (b3). For the P4: C18H9ON2S2-OCH3 has link lengths in the following ranges: C–H of 1.101 Å ÷ 1.103 Å, C–N of 2.709 Å, C–S of 1.452 Å ÷ 1.470 Å, C–C of 1.504 Å ÷ 1.532 Å, C–O of 1.440 Å ÷ 1.454 Å, and the following link angle between the atoms as H–C–H of 106.548° ÷ 106.711°, H–C–C of 110.285° ÷ 110.927°, C–C–C of 109.631° ÷ 109.842°, H–C–N of 109.434° ÷ 109.770°, C–S–C of 106.609° ÷ 111.975°, C–N–C of 89.924°, C–O–C of 104.513° ÷ 113.274° (a4). For the P5: C18H9ON2S2-Cl has link lengths in the following ranges: C–H of 1.106 Å ÷ 1.107 Å, C–N of 1.444 Å ÷ 1.451 Å, C–S of 1.890 Å ÷ 1.917 Å, C–C of 1.519 Å ÷ 1.540 Å, C–O of 1.451 Å ÷ 1.507 Å, and the following link angles between the atoms as H–C–H of 106.966° ÷ 108.930°, H–C–C of 109.027° ÷ 109.918°, C–C–C of 110.002° ÷ 112.362°, H–C–N of 104.437° ÷ 106.069°, C–S–C of 105.776° ÷ 111.053°, C–N–C of 111.664°, C–O–C of 107.799°, Cl–C–C of 110.442° (a5). The obtained results indicated that the link lengths between the atoms in the P1, P2, P3, P4, and P5 have values changed significantly with only the link length C–S of 1.880 Å ÷ 1.994 Å for the P1, C–O of 1.451 Å ÷ 1.492 Å for the P2, C–O of 1.459 Å ÷ 1.483 Å for the P3, C-O of 1.440 Å ÷ 1.454 Å for the P4, C–Cl of 1.873 Å ÷ 1.874 Å for the P5. The change in link lengths is due to the electrostatic interaction when the O atom in P1 is exchanged by S (in P2) and the H atom in P3 is exchanged by OCH3 (in P4) and Cl (in P5). This factor has led to a decrease in the total energy of the system (Etot) corresponding to the polymers as P1: Etot of Etot = – 1904 eV, P2: Etot of Etot = – 2548 eV, P3: Etot of Etot = – 3348 eV, P4: Etot of Etot = – 3575 eV, P5: Etot of Etot = – 4264 eV. Besides, the band gap (Eg) corresponds to the P1: C11H7NS-O has Eg of Eg = 0.6210 eV (b1). When replacing – O by – S, the structural shape has the form of P2: C11H7NS-S (a2) and Eg of Eg = 0.2390 eV (b2). Similarly, the P3, C18H9ON2S2-H has the Eg of Eg = 0.0060 eV (b3) when replacing – H by – OCH3, the P4, C18H9ON2S2-OCH3 has the Eg of Eg = 0.0640 eV (b4), – H equals – Cl in the P5, C18H9ON2S2-Cl with Eg of Eg = 0.0645 eV (b5). These results indicated that when replacing O with S, the Eg decreased, and replacing the O atom in P1 with S led to the decrease of Etot and Eg, and with – H of P3 by – OCH3, – Cl, then Eg increased, and Etot decreased. When a benzo[d]thiazole was added in P1 (changing into P3), the Eg was extremely strongly decreased, nearly 100 times (from Eg = 0.6210 eV to Eg = 0.0060 eV). The results obtained are very helpful for these future experimental researches. To confirm, we have studied the electron density in the energy bands. The results of electron density in the energy bands of C11H7NS with different impurities are shown in .
Figure 4. Electron density in the energy bands (E) of P1, P2, P3, P4, and P5
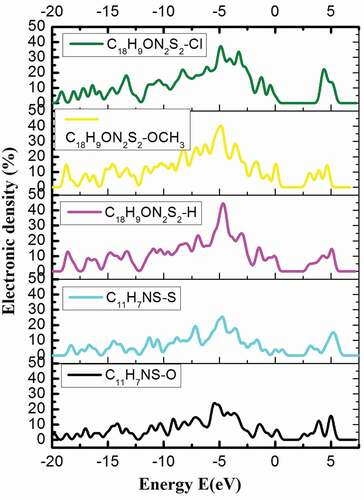
The obtained results show that the electronic density of P1: C11H7NS-O, P3: C18H9ON2S2-H with the energy bands (E) of E = – 20 eV, – 15 eV, – 10 eV, – 7.5 eV, – 5 eV, 0.00 eV, 5 eV, or 7.5 eV has the corresponding electronic density P1: 0.00%, 4.55%, 2.69%, 18.76%, 23.66%, 3.96%, 13.80%, 0.00%; P3: 0.00%, 10.98%, 11.57%, 17.29%, 36.84%, 9.71%, 14.45%, 0.00%. When doping the functional groups – S, – OCH3, or – Cl into P1 and P3, we obtained P2, P4, and P5, which showed significant changes in electron density. For example, in the E = – 20 eV energy range, the electron density has increased from 0.00% to 2.55% then back to 0.00%; in the energy range of E = – 15 eV, the electron density decreases and then increases and vice versa from 4.55% to 4.41%, up 10.98%, 11.82%, down to 6.26%; in the energy range of E = – 10 eV, the electron density increases and then decreases from 2.69% to 7.34%, 11.57%, 14.58%, down to 12.05%; at the energy range of – 7.25 eV, the electron density change to reach the extreme value in the valence area, tends to change from 18.76% to 14.57% to 17.29%, 22.13%, to 17.21%; in the E = −5 eV energy range, the density of electrocytes change from 23.66% to 22.27% to 36.84%, 39.75%, 36.15%; in the range with E = 0.00 eV the electronic density increased and then decreased from 3.96% to 5.80%, 9.71%, 14.94%, 4.83%; in the energy range of E = 5 eV, the electron density increased and then decreased from 13.80% to 15.62%, down to 14.45%, 3.55% to 13.80%; in the energy range E = 7.25 eV, the polymer has an electron density of 0.00%. This showed that in the electron density, the valence region had the maximum value, the largest percentage in the energy range of E = – 5 eV (). Through the obtained calculations, we confirm that this is still a semiconductor material and conductivity was increased because doping the -S functional group into P1 leads to band gap Eg decrease and the conductivity increase. Similarly, doping the functional groups – OCH3, or – Cl into P3 leads to an increase in the band gap of Eg. Besides, connecting P1 and P3 through the benzene bridge results in a huge decrease of Eg nearly 100 times, while Etot decreased nearly 2 times. The results are shown in the first Brillouin region (corresponding to zero) correspond to an increase in electron density from 3.96% to 5.80%, 9.71%, 14.94%, and then to 4.83% that increase in electrical mobility. The electrical mobility of P1 decreased when it was doped with -S, and increased when – OCH3 or – Cl was added to P3. It confirmed that the impurities affected the network structure and electronic structure of P1 and P3. The cause of this phenomenon is due to the influence of the electronic structure of the functional groups on the band gap Eg and the total energy of the system Etot. The obtained results are very useful for future experimental results used to fabricate smart electronic device.
4. Conclusions
This paper summarizes the structural calculations and gives a detailed comparison of the lattice structure, and electronic structure of C11H7NS-X and C18H9ON2S2-R in case of X and R was replaced with other elements or groups. The change in link length, link angle, total energy, and band gap of doped polythiophenes is convincing evidence to confirm the influence of the impurities, the benzene ring on the band gap, and electronic structure features of polymers. Among investigated dopants, the benzo[d]thiazole ring exhibits the strongest effect on the band gap polymers. These results are considered the basis for future experimental work related to doping on the polythiophenes containing benzo[d]thiazole and benzo[d]oxazole and used to fabricate smart electronic device.
Acknowledgments
The authors thank the Hanoi National University of Education for the supports of the computer system.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- Ikai T, Takayama K, Wada Y, et al. Synthesis of a one-handed helical polythiophene: a new approach using an axially chiral bithiophene with a fixed syn-conformation. Chem Sci. 2019;10(18):4890–4895.
- Valderrama-García B, Rodríguez-Alba E, Morales-Espinoza E, et al. Synthesis and characterization of novel polythiophenes containing pyrene chromophores: thermal, optical and electrochemical properties. Molecules. 2016;21(2):172.
- Pina J, Beltran Rodrigues AC, Alnady MMSA, et al. Restricted aggregate formation on tetraphenylethene-substituted polythiophenes. J Phys Chem C. 2020;124(25):13956–13965.
- Gupta S, Chatterjee S, Zolnierczuk P, et al. Impact of local stiffness on entropy driven microscopic dynamics of polythiophene. Sci Rep. 2020;10(1):9966.
- Langeveld-Voss BMW, Janssen RAJ, Meijer EW. On the origin of optical activity in polythiophenes. J Mol Struct. 2000;521(1–3):285–301.
- Kaloni TP, Giesbrecht PK, Schreckenbach G, et al. Polythiophene: from fundamental perspectives to applications. Chem Mater. 2017;29(24):10248–10283.
- Hu Z, Zhang S, Zhang C, et al. Donor–acceptor units modulate the electronic and photoluminescence characteristics of thiophene oligomers. J Appl Phys. 2019;126(24):245501.
- Nguyen NL, Tran TTD, Nguyen H, et al. Synthesis of polythiophene containing heterocycle on the side chain: a review. Vietnam J. Chem. 2020;58(1):1–9.
- Djemoui A, Naouri A, Ouahrani MR, et al. A step-by-step synthesis of triazole-benzimidazole-chalcone hybrids: anticancer activity in human cells+. J Mol Struct. 2020;1204:127487.
- Tokárová Z, Maxianová P, Váry T, et al. Thiophene-centered azomethines: structure, photophysical and electronic properties. J Mol Struct. 2020;1204:127492.
- Vu QT, Tran TTD, Dang TT, et al. Some chalcones derived from thiophene-3-carbaldehyde: synthesis and crystal structures. Acta Cryst. Section E. 2019;75(7):957–963.
- Esquivel ECC, Rufino VC, Nogueira MHT, et al. Synthesis and characterization of 1,3,5-triarylpyrazol-4-ols and 3,5-diarylisoxazol-4-ols from chalcones and theoretical studies of the stability of pyrazol-4-ol toward acid dehydration. J Mol Struct. 2020;1204:127536.
- Levesque I, Bazinet P, Roovers J. Optical properties and dual electrical and ionic conductivity in Poly(3-methylhexa(oxyethylene)oxy-4-methylthiophene). Macromolecules. 2000;33(8):2952.
- Ming S, Youjun H, Kunlun H, et al. A water-soluble polythiophene for organic field-effect transistors. Polym Chem. 2013;4(20):5270–5274.
- Pei J, Yu WL, Ni J, et al. Thiophene-based conjugated polymers for light-emitting diodes: effect of aryl groups on photoluminescence efficiency and redox behavior. Macromolecule. 2001;34(21):7241.
- Snook GA, Kao P, Best AS. Conducting-polymer-based supercapacitor devices and electrodes. J Power Sources. 2011;196(1):1–12.
- Ghaitaoui T, Ali B, Sahli Y, et al. Realization and characterization of p-typed polythiophene based organic photovoltaic cells. Journal of Nano- and Electronic Physics. 2018;10(1):01008.
- Nabasmita M, Radhakanta G, Arun Nandi K. Optoelectronic properties of self-assembled nanostructures of polymer functionalized polythiophene and graphene. Langmuir. 2018;34(26):7585–7597.
- Borrelli DC, Lee S, Gleason KK. Optoelectronic properties of polythiophene thin films and organic TFTs fabricated by oxidative chemical vapor deposition. Journal of Materials Chemistry C. 2014;2(35):7223–7231.
- Sandip D, Dhruba PC, Radhakanta G, et al. Water soluble polythiophenes: preparation and applications. RSC Adv. 2015;5(26):20160–20177.
- Maiti J, Pokhrel B, Boruah R, et al. Polythiophene based fluorescence sensors for acids and metal ions. Sens Actuators B Chem. 2009;141(2):447–451.
- Wang F, Li M, Wang B, et al. Synthesis and characterization of water-soluble polythiophene derivatives for cell imaging. Sci Rep. 2015;5(1):7617.
- Aldo VA, Gerardo ZG, Edgar AO, et al. Luminescent polythiophenes-containing porphyrin units: synthesis, characterization, and optical properties. Designed Monomers & Polymers. 2014;17(1):78–88.
- Kambiz S, Jeong-Yeol Y, Jongchul S, et al. Chromogenic polymers and their packaging applications: a review. Polymer Rev. 2020;60(3):442–492.
- Elbing M, Garcia A, Urban S, et al. In Situ conjugated polyelectrolyte formation. Macromolecules. 2008;41(23):9146–9155.
- Aiello S, Wells G, Stone EL, et al. Synthesis and biological properties of benzothiazole, benzoxazole, and chromen-4-one analogues of the potent antitumor agent 2-(3,4-dimethoxyphenyl)-5-fluorobenzothiazole (PMX 610, NSC 721648). J Med Chem. 2008;51(16):5135–5139.
- Cho Y, Ioerger TR, Sacchettini JC. Discovery of novel nitrobenzothiazole inhibitors for mycobacterium tuberculosis ATP phosphoribosyl transferase (HisG) through virtual screening. J Med Chem. 2008;51(19):5984–5992.
- Radhakrishnan S, Parthasarathi R, Subramanian V, et al. Structure and properties of polythiophene containing hetero aromatic side chains. Comput Mater Sci. 2006;37(3):318–322.
- Radhakrishnan S, Somanathan N. Poly(thiophenes) functionalised with thiazole heterocycles as electroluminescent polymers. J Mater Chem. 2006;16(29):2990–3000.
- Radhakrishnan S, Somanathan N, Thelakkat M. Thermal degradation studies of polythiophenes containing hetero aromatic side chains. Int J Thermophys. 2009;30(3):1074–1108.
- Esashika K, Yoshizawa-Fujita M, Takeoka Y, et al. Synthesis and optical properties of poly(thiophene-fluorene) copolymers with benzothiazole moiety. Synth Met. 2009;159(21–22):2184–2187.
- Nguyen NL, Vu QT, Duong QH, et al. Green synthesis and crystal structure of 3-(benzo-thia-zol-2-yl)thio-phene, Acta crystallographica. Section E, Crystallographic communications. 2017;73(11):1647–1651.
- Radhakrishnan S, Parthasarathi R, Subramanian V, et al. Quantum chemical studies on polythiophenes containing heterocyclic substituents: effect of structure on the band gap. J Chem Phys. 2005;123(16):164905.
- Thaneshwor PK, Georg S, Michael SF. Band gap modulation in polythiophene and polypyrrole-based systems. Sci Rep. 2016;6(1):36554.
- Arnold CA, Wilfredo CC, Rolando VB, et al. AB initio and density functional studies of polythiophene energy band gap. NECTEC Technical Journal. 2001;9:215–218.
- Brocks G. Density functional study of polythiophene derivatives. J Phys Chem. 1996;100(43):17327–17333.
- Si MB, Guillermo SM, Mohamed H, et al. DFT study of polythiophene energy band gap and substitution effects. J Chem. 2015;9:12. Article ID 296386
- Ali SR, Mehri E, Etesam G, et al. The polythiophene molecular segment as a sensor model for H2O, HCN, NH3, SO3, and H2S: a density functional theory study. J Mol Model. 2016;22(6):127. 8
- Anusuya S, Bishwajit G. A DFT study to probe homo-conjugated norbornylogous bridged spacers in dye-sensitized solar cells: an approach to suppressing agglomeration of dye molecules. RSC Adv. 2020;10(26):15307–15319.
- Nguyen NH, Ngo TC, Hoang VH, et al. Electronic properties of the polypyrrole-dopant anions <sub>ClO4 − and <sub>MoO42−: a density functional theory study. J Mol Model. 2017;23(12):336.
- Muhammad AA, Shaikh M, Fatma K, et al. Synthesis and characterization of poly(3-hexylthiophene): improvement of regioregularity and energy band gap. RSC Adv. 2018;8(15):8319–8328.
- Vu QT, Tran TTD, Nguyen TC, et al. DFT prediction of factors affecting the structural characteristics, the transition temperature and the electronic density of some new conjugated polymers. Polymers. 2020;12(6):1207.
- Rittmeyer SP, Gro A. Structural and electronic properties of oligo- and polythiophenes modified by substituents, Beilstein J. Nanotechnol 2012;3:909–919.
- Banjo S, Ayobami OO, Ajibade AI. Structural and electronic properties of 4H-cyclopenta[2,1-b,3;4-b′]dithiophene S-oxide (BTO) derivatives with an S, S=O, O, SiH2, or BH2 bridge: semi-empirical and DFT study. J Mol Model. 2011;18(6):2755–2760.
- Lo PK, Lau KC. High-level ab initio predictions for the ionization energies and heats of formation of five-membered-ring molecules: thiophene, Furan, Pyrrole, 1,3-Cyclopentadiene, and Borole, C4H4X/C4H4X+(X = S, O, NH, CH2, and BH). J Phys Chem A. 2011;115(5):932–939.
- Barlow S, Odom SA, Lancaster K, et al. Electronic and optical properties of 4H-Cyclopenta[2,1-b:3,4-b′]bithiophene derivatives and their 4-Heteroatom-substituted analogues: a joint theoretical and experimental comparison. J Phys Chem A. 2010;114(45):14397–14407.
- Kien PH, Lan MT, Dung NT, et al. Annealing study of amorphous bulk and nanoparticle iron using molecular dynamics simulation. Int J Modern Phys B. 2014;28(23):1450155. 17
- Trong DN, Chinh CN, The TN, et al. Factors on the magnetic properties of the iron nanoparticles by classical heisenberg model. Physica B. 2018;532:144–148.
- Nguyen-Trong D, Nguyen-Tri P. Understanding the heterogeneous kinetics of Al nanoparticles by simulations method. J Mol Struct. 2020;1218(9):128498.
- Quoc TT, Trong DN. Molecular dynamics factors affecting on the structure, phase transition of Al bulk. Phys B Condens Matter. 2019;570:116–121.
- Quoc TT, Trong DN, tefan S¸, et al. Study on the influence of factors on the structure and mechanical properties of amorphous aluminium by molecular dynamics method. Adv Mater Sci Eng. 2021;2021(10):5564644.
- Nguyen TD, Nguyen CC, Hung Tran V. Molecular dynamics study of microscopic structures, phase transitions and dynamic crystallization in Ni nanoparticles. RSC Adv. 2017;7(41):25406–25413.
- NguyenTrong D. Z-AXIS deformation method to investigate the influence of system size, structure phase transition on mechanical properties of bulk nickel. Mater Chem Phys. 2020;252(6):123275.
- Dang Thi Minh H, Coman G, Nguyen Quang H, et al. Influence of heating rate, temperature, pressure on the structure, and phase transition of amorphous Ni material: a molecular dynamics study. Heliyon. 2020;6(11):e05548.
- Trong DN, Chinh CN, Quoc VD, and Tuan Tran Quoc. Study the effects of factors on the structure and phase transition of bulk Ag by molecular dynamics method. International Journal of Computational Materials Science and Engineering. 11. 2020;9(3):2050016.
- Nguyen-Trong D, Nguyen-Tri P. Factors affecting the structure, phase transition and crystallization process of AlNi nanoparticles. J Alloys Compd. 2020;812(8):152133.
- Nguyen-Trong D, Nguyen-Tri P. Molecular dynamic study on factors influencing the structure, phase transition and crystallization process of NiCu6912 nanoparticle. Mater Chem Phys. 2020;250(6):123075.
- Tuan TQ, Dung NT. Effect of heating rate, impurity concentration of Cu, atomic number, temperatures, time annealing temperature on the structure, crystallization temperature and crystallization process of Ni1-xCux bulk; x = 0.1, 0.3, 0.5, 0.7”. Int J Modern Phys B. 2018;32(26):1830009. 15
- Nguyen-Trong D, Pham-Huu K, Nguyen-Tri P. Simulation on the factors affecting the crystallization process of FeNi alloy by molecular dynamics. ACS Omega. 2019;4(11):14605–14612.
- Trong Dung N. Influence of impurity concentration, atomic number, temperature and tempering time on microstructure and phase transformation of Ni1−xFex (x= 0.1, 0.3, 0.5) nanoparticles. Mod Phys Lett B. 2018;32(18):1850204. 14
- Van Cao L, Van Duong Q, Nguyen Trong D. Ab Initio Calculations on the structural and electronic properties of AgAu Alloys. ACS Omega. 2020;5(48):31391–31397.
- Trong DN, Long VC, Ţălu S. The structure and crystal-lizing process of NiAu alloy: a molecular dynamics simulation method. J Composites Sci. 2021;5(1):1–14.
- Quoc TV, La Trieu D, Van Duong Q, et al. Effect of doped H, Br, Cu, Kr, Ge, As and Fe on structural features and bandgap of poly C13H8OS-X: a DFT calculation. Des Monomers Polym. 2021;24(1):53–62.
- Arjunan V, Thirunarayanan S, Durga DG, et al. Substituent influence on the structural, vibrational and electronic properties of 2,5-dihydrothiophene-1,1-dioxide by experimental and DFT methods. Spectrochim Acta A Mol Biomol Spectrosc. 2015;150:641–651.
- Yılmaz ZT, Yasin Odabaşoğlu H, Şenel P, et al. A novel 3-((5-methylpyridin-2-yl)amino)isobenzofuran-1(3H)-one: molecular structure describe, X-ray diffractions and DFT calculations, antioxidant activity, DNA binding and molecular docking studies. J Mol Struct. 2020;1205:127585.
- Prabavathi N, Senthil NN, Venkatram RB. Molecular structure, vibrational spectra, natural bond orbital and thermodynamic analysis of 3,6-dichloro-4-methylpyridazine and 3,6-dichloropyridazine-4-carboxylic acid by DFT approach. Spectrochim Acta A Mol Biomol Spectrosc. 2015;136:1134–1148.
- Kantchev EAB, Norsten TB, Tan MLY, et al. Thiophene-containing pechmann dyes and related compounds: synthesis, and experimental and DFT characterisation. Chem A Eur J. 2011;18(2):695–708.
- Thaneshwor PK, Georg S, Michael SF. Band gap modulation in polythiophene and polypyrrole-based systems. Sci Rep. 2016;6(1):36554.
- Fréchette M, Belletete M, Bergeron J-Y, et al. Monomer reactivity vs regioregularity in polythiophene derivatives: a joint synthetic and theoretical investigation. Synth Met. 1997;84(1–3):223–224.
- Sun H, Autschbach J. Electronic energy gaps for π-conjugated oligomers and polymers calculated with density functional theory. J Chem Theory Comput. 2014;10(3):1035–1047.
- Hussain M, Arshad N, Ujan R, et al. Synthesis, structure elucidation and surface analysis of a new single crystal N-((2-(benzo [4,5]imidazo [1,2-c]quinazolin-6-yl)phenyl)carbamothioyl)heptanamide: theoretical and experimental DNA binding studies. J Mol Struct. 2020;1205:127496.
- Delley B. An all-electron numerical method for solving the local density functional for polyatomic molecules. J Chem Phys. 1990;92(1):508–517.
- Delley B. From molecules to solids with the DMol3 approach. J Chem Phys. 2000;113(18):7756–7764.
- Perdew JP, Burke K, Ernzerhof M. Generalized gradient approximation made simple. Phys Rev Lett. 1996;77(18):3865–3868.
- Tsuzuki S, Lüthi HP. Interaction energies of van der Waals and hydrogen bonded systems calculated using density functional theory: assessing the PW91 model. J Chem Phys. 2001;114(9):3949.
- Perdew JP, Wang Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys Rev B. 1992;45(23):13244–13249.
- Monkhorst HJ, Pack JD. Special points for Brillouin-zone integrations. Phys Rev B. 1976;13(12):5188–5192.