
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Density functional theory (DFT) and time-dependent DFT (TD-DFT) are used to investigate the ground- and excited-state properties of donor-acceptor–donor (D-A-D) monomers based on 3,6-carbazole (CB) combined with various-conjugated benzothiazole derivatives, using B3LYP and the 6–311 G basis set. To create nine D-A-D monomers for this investigation, nine (9) distinct acceptors were inserted at the C3 and C6 positions of carbazole. The impact of various electron-donor groups on structural, electrical, and optoelectronic properties is investigated. Our technique for developing novel donor monomers provides a theoretical framework for further optimizing the photovoltaic device’s electrical, optical, and efficiency features. The HOMO and LUMO energies, bandgap, excited state, exciton binding energy, open-circuit voltage (VOC) and absorption spectra were calculated. Our findings indicate that CB-TDP-CB and CB-SDP-CB monomers have an appropriate electronic structure for polymer solar cells.
1. Introduction
Solar energy collecting is one of the most challenging issues facing today’s renewable energy specialists. Researchers are working hard to develop low-cost, dependable, elastic, and ecologically friendly optoelectronic devices with high efficiency [Citation1–4]. Although silicon-based materials are appropriate for current solar cell technology, their high cost and high operating temperature limit their portability and adaptability [Citation5]. Conjugated polymers (CPs) were used as a step toward the creation of low-cost, environmentally friendly, easily synthesizable, flexible, and efficient materials for solar cells [Citation4,Citation6]. Because of their stability, low-cost manufacturing, and ability to build tunable and robust structures, CPs is emerging as attractive materials. Scientists are investigating four prominent generations of CPs, which have applications in solar to electrical energy conversion [Citation7–11].
Conjugated polymers have sparked significant interest in recent decades [Citation12,Citation13] due to their potential applications in fields such as organic field-effect transistors [Citation14], organic light-emitting diodes [Citation15], electrochromic devices [Citation16], photovoltaics [Citation17], electronic displays, supercapacitors, thermoelectric devices [Citation18], and so on.
Conjugated polymers with a donor-acceptor donor (D-A-D) backbone are promising candidates for organic semiconductors because they allow for the customization of optoelectronic properties by modifying the donor and acceptor units. A wide range of electron-rich and electron-deficient polymers can be synthesized by carefully selecting donor and acceptor units, resulting in a wide range of electron-rich and electron-deficient polymers for electron and hole stabilization with fine bandgap control and energy levels as needed. The copolymerization of D-A-D monomer with other building blocks can more effectively modify the optoelectronic properties of D-A-D polymer. Copolymers have recently gained much attention because of their tunable physical and chemical properties, such as bandgap, highest occupied molecular orbital (HOMO), lowest unoccupied molecular orbital (LUMO), optical absorption, shape, solubility, stability, and so on [Citation19]. It should also be mentioned that a wide range of donor and acceptor units are now available for fine-tuning the optoelectronic properties [Citation20].
D-A-D plays a vital function in charge separation and molecular architecture, which regulates charge transfer. The D-A-D system exhibits reduced band gap, significant charge transformation, and improved visible light absorption due to considerable overlapping of D and A molecular orbitals. Therefore, one of the criteria for generating a large photocurrent has been the synthesis of donor and acceptor materials with complementary absorption characteristics intended to increase the coverage of the solar spectrum. As a result, for OPVs, a range of narrow optical gap non-fullerene acceptors (NFAs) based on a stronger electron-donating core were carefully designed and synthesized [Citation21]. Furthermore, utilizing a D-A-D fused core can choose a somewhat planar structure rather than a twisted one, facilitating electron flow from donor to acceptor since the planarized D-A-D promotes -electrons delocalization. More crucially, a modest electron perturbation may occur in the D-A-D fused core, resulting in efficient charge transfer [Citation22]. In other words, when the triazole is introduced into this fused system, -orbital electrons are less likely to be caught while travelling through the fused -bridge [Citation23].
The current study’s novelty stems from the usage of new donor-acceptor-donor monomers as a 3,6- carbazole donor and benzothiazole-based derivates as acceptors, based on these preliminary findings and theoretical models. Numerous studies show alternating polymeric architectures to identify the best poly(3,6-carbazole) derivatives for BHJ solar cell devices [Citation24–27]. Quantum calculations were performed on the repeat unit of the proposed polymers to determine both the HOMO and LUMO energy levels to evaluate the poly-carbazole performances utilizing those models. These novel polymers were put through their paces in solar cell devices, and their performance was assessed in terms of polymer organization, molecular weight, charge carrier mobility, and LUMO energy level.
Carbazol-based polymers (PCz) have gotten much interest in the last 50 years because they are more stable and have a more significant redox potential than other conducting polymers [Citation28]. Similarly, their excellent hole transportation mobility and substantial absorption in the UV spectral region have good electro- and photoactive characteristics [Citation29]. It is undeniably a successful method to raise the HOMO level while decreasing the optical gap. Standard building blocks for boosting a single component’s photoluminescence quantum yield (PLQY) are benzothiazole-based conjugated compounds with distinctive luminescence characteristics [Citation30]. Furthermore, the strong PLQY indicates efficient radioactive recombination pathways, leading to a high electroluminescence yield of the final devices. As a result, it aims to insert benzothiazole into the central core to construct an electron-deficient-core-based fused structure (D-A-D) for altering the resulting molecules’ optoelectronic properties to achieve low voltage loss and high device performance.
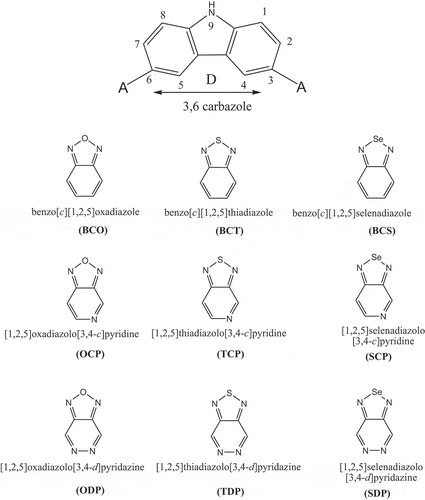
In this investigation, D-A-D monomers 3,6-carbazoles were utilized as donors (D) and accepters such as: benzо[с] [1,2,5] оxаdiаzоle (BСО); benzо[с] [1,2,5] thiаdiаzоle (BСT); benzо[с][1,2,5]selenаdiаzоle (BСS); [1,2,5] оxаdiаzоlо[3,4-с]рyridine (ОСР); [1,2,5] thiаdiаzоlо [3,4-с]рyridine (TСР); [1,2,5] selenаdiаzоlо [3,4-с] рyridine (SСР); [1,2,5] оxаdiаzоlо[3,4-d]рyridаzine (ОDР); [1,2,5] thiаdiаzоlо[3,4-d]рyridаzine (TDР) аnd [1,2,5]selenаdiаzоlо[3,4-d]рyridаzine (SDР) ().
Because of high-performance computing and optimization of computational chemistry programs, theoretical investigations in this area have increased in recent years. The theoretical method is the most effective instrument for overcoming difficulties in experimental synthesis and exploring alternatives that lower material production and processing costs. Because DFT investigates this type of material’s electronic structure and spectroscopic properties, it represents a reliable alternative for tackling these jobs. We created a new D-A-D type of small-molecule OPV donor in this study. Because there are so many conceivable D and A unit combinations, a virtual pre-synthesis screening would be required for a logical design of small-molecule OPV donors. With density functional theory (DFT) and time-dependent DFT (TDDFT) calculations at the B3LYP/6-31 G level, we have well defined molecular orbital (MO) energy levels, bandgaps, UV/VIS absorption spectra, and PCEs of diverse D-A-D polymer donors.
2. Computational calculation details
The same calculation method as in our prior investigations [Citation31,Citation32] is used. The DFT approach of Becke’s three-parameter compound (B3LYP) [Citation33–35] was employed in all of the neutral monomer studies in this work. All computations were performed using the 6–311 G basis set. The Gaussian09 package [Citation36] was used to perform the density (DFT) and time-dependent (TD-DFT) functional calculations [Citation37]. We determined the HOMO and LUMO energies of the molecules analyzed using optimal structures; we also determined the bandgap energy (Eg), which was calculated as the difference between the HOMO and LUMO energy levels. At the TD-DFT/B3LYP/6-31 G level, the absorption spectra and excited states of all the monomers (D-A-D) structures were computed. The Gausssum software tool was also used to visualize the total density of states (TDOS).
Because most chemical activities occur in the solution phase, solvent effects garner a lot of interest. Therefore, the solvent effects are included here to guarantee that the calculations are compatible with the common experimental settings. The following section goes into greater detail about the impact of the chemical environment. To investigate the effect of solvents on ground-state molecular geometry, quantum chemical calculations of excitation energies and absorption maxima on the examined molecules were performed in both vacuum and solvent. In the TD-DFT calculations, chlorobenzene was employed as the solvent within the polarizable continuum model (PCM) [Citation38].
3. Results and discussions
3.1. Molecular design and geometry structures
The structural properties of the nine D-A-D monomers examined are depicted in Fig. S1, and their optimal geometries are displayed in . The molecules are completely optimized in the neutral states to establish the geometric parameters; we used the 6–311 G basis set. We found that the geometric characteristics are slightly affected by the different acceptor groups connected to the donor molecule. shows the inter-cyclic lengths and dihedral angles between D-A in D-A-D monomer determined from the optimized structures of the examined molecules in the neutral state in gas and solvent. Planarity in molecule geometry and the corresponding -electrons conjugation over the backbone influence a chemical substance’s visible light absorption. An ideal 1800 dihedral of the D-A-D () demonstrates that the combination of donor and acceptor moieties planarized the geometry of the resultant polymer by producing a delocalized -electronic cloud density across the backbone. The D-A-intra-chain D’s dihedral angle (1800) is responsible for its low cost and simple synthesis technique.
Figure 1. Optimized geometries of the studied molecules obtained by B3LYP/6-311 G in the gas phase
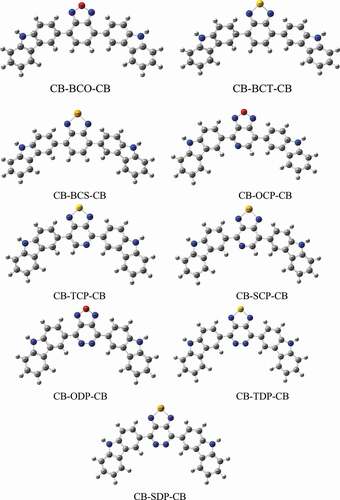
Figure 2. Representation of the frontier molecular orbitals (HOMO and LUMO) obtained from DFT//B3LYP/6-31 G+(d,p) calculations in the gas phase
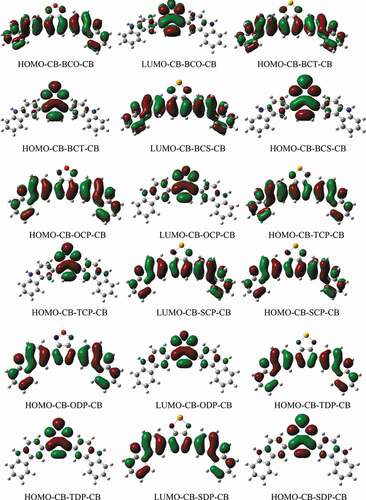
Figure 3. The HOMO and LUMO energy levels of the studied D-A-D monomers at DFT/ B3LYPlevel with 6–311 G basis set
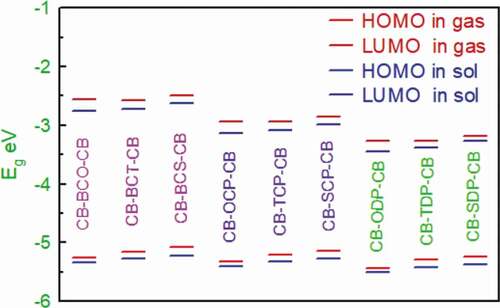
Figure 4. Simulated UV-Visible optical absorption spectra of the studied carbazole copolymer monomers (D-A-D) calculated by TD/DFT/ B3LYP/6-311 G level in the gas phase
Figure 5. Simulated UV-Visible optical absorption spectra of the studied carbazole copolymer monomers (D-A-D) calculated by TD/DFT/ B3LYP/6-311 G level in the solvent phase
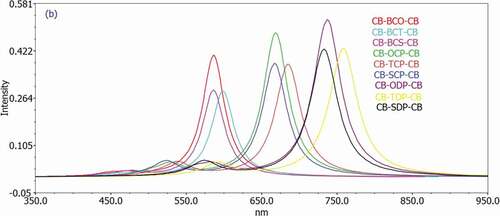
Figure 6. The density of states (DOS) diagram produced by GaussSum for Studied D-A-D monomers
Table 1. Intercyclic distances (Å) and dihedral angles between D-A unit’s values of the studied monomers obtained in the gas and solvent phase by using DFT/B3LYP with 6–311 G basis set
According to the findings in , the dihedral angles in the intramolecular rings of D-A-D are likewise close to planarity. We discovered a modest effect of the acceptor group on the inter-cyclic distance, but the impact on the dihedral angles θi° (i = 1–9) is visible in both the gas and solvent phases. We also said that the solvent phase affects these factors. In reality, the inter-cyclic lengths decrease while the dihedral angles increase significantly, allowing for improved electrical conjugation. In the monomer models, the two units are twisted from each other in the minimum energy structures of 1, 2, and 5 by 300–380, most likely due to repulsion between the two hydrogen atoms projecting from the phenyl groups present in both units. The repulsion was removed by substituting a benzothiazole for the phenyl group in the fused-ring unit; hence, 4,6,7,8 and 9 have almost planar lowest energy structures.
3.2. Electronic prosperities
3.2.1. Frontier molecular orbitals (FMOs)
FMOs are one of the most underlying molecular orbitals because of their association with chemical species reactivity. The FMOs in PSCs are critical for understanding and predicting how charge transfer throughout the polymer occurs due to an excitation event. The HOMO and LUMO diagrams of the D-A-D monomers are shown in . The HOMO density is spread along with the structure for all D-A-D monomers, as seen in . Furthermore, this FMO is found in the C-C bonds of the carbazole donor group. The density of the LUMO, on the other hand, is primarily distributed among the acceptor groups. As a result of their excellent superposition, the current PSC candidates provide an ideal conduit for electron transport from the HOMO to LUMO orbital.
Predicting the behaviour of new polymers’ HOMO and LUMO energy levels is critical for rationally designing optimum BHJ solar cells. We computed the HOMO and LUMO energy levels of the D-A-D monomers using density functional theory (DFT) as represented by the B3LYP functional and the 6–311 G basis set in this work. The DFT/B3LYP/6-311 G formalism has been proven to be an accurate framework for determining numerous molecular systems’ structural and optical properties [Citation39]. The HOMO and LUMO energy levels and the bandgaps of the copolymers are directly related to the efficiency of solar cells.
The electrical properties were utilized to anticipate the primary qualities useful in polymer solar cells. shows some of the parameters found via DFT calculations. depicts the FMO energy levels and energy gaps of the all monomers model (n = 1–9). In addition, the HOMO energy levels of the 1–9 monomers in the gas phase and solvent phase are presented in , implying that these systems may have strong air stability when applied to BHJSC with PC60BM. Indeed, a statistical review [Citation40] proposes that within a photovoltaic device employing PC60BM as an acceptor, a lower HOMO level (closer to vacuum) of the polymer donor will aid improve open-circuit voltage (VOC) devices.
Table 2. Calculated EHOMO, ELUMO levels, energy gap (Eg) values of the studied monomers obtained by DFT/B3LYP/6-311 G G level
shows the HOMO-LUMO energy gaps (Eg = ELUMO – EHOMO) for D-A-D monomers. Except for the CB-SCP-CB monomer, the bandgap (Eg) values are less than 3 eV. CB-TDP-CB> CB-SDP-CB> CB-ODP-CB> CB-ODP-CB> CB-TCP-CB> CB-OCP-CB is the order of D-A-D monomers concerning Eg. In both phases, CB-BCS-CB CB-BCT-CB CB-BCO-CB CB-SCP-CB The lower Eg of CB-TDP-CB and CB-SDP-CB, compared to other monomers, illustrates the importance of intramolecular charge movement, which causes the ingestion spectra to redshift. This is clearly due to the impact of the electron-contributor unit, which is more solid for 3,6-CB-TDP-CB and 3,6-CB-SDP-CB than for other monomers. Therefore, all atoms with a minimal energy gap must have extraordinary photophysical properties, particularly 3,6-CB-TDP-CB.
This suggests that the degree of inductive effects of the acceptor units considerably influences the band gaps of the constructed molecules. Furthermore, these findings indicated that adding sulfur, nitrogen, and oxygen groups help close energy gaps. In that scenario, the CB-SCP-CB monomer exhibits the most significant energy gap and a drop in the HOMO level, which should be directly related to the increased electron-donating capability of the SCP acceptor unit containing the Se atom. These findings indicate that acceptor units have a significant impact on the LUMO distribution and, as a result, the HOMO-LUMO energy gaps. It is proposed that the disparity in the distribution of the LUMO orbital for the designed systems is due to the considerable variance in their geometric qualities, as 3,6-CB-TDP-CB and 3,6-CB-SDP-CB are the most planar and hence the most conjugated.
3.3. Photovoltaic properties
3.3.1. Scharber’s model
The device’s maximum power density divided by the total power density received from the Air Mass 1.5 solar spectrum [Citation41], which is 1000 W/m2. The power density of the device is made up of the open-circuit voltage, short-circuit current density, and fill factor (FF). According to Scharber’s model, the VOC is associated with the gap between the acceptor’s LUMO and the donor’s HOMO. Subtracting 0.3 eV from the energy level difference yields the VOC. This change was identified empirically and is associated with residual carrier binding energy and interface effects [Citation42]. External quantum efficiency (EQE) multiplied by the number of photons from the Air Mass 1.5 sun spectrum at all frequencies. The EQE is just a step function with a value of 0 per cent for energies below and above the donor’s optical band gap (Eopt). For all devices, the fill factor is FF = 0.65. If needed, further EQE and FF assumptions can be made. The EQE, for example, could be found by analyzing the Kohn-Sham joint density of states, which reveals the frequency-dependent absorption cross-section behaviour. In this case, the polymer layer is thick enough to absorb any photon passing across the optical band gap, while the film structure effectively restricts the EQE. Scharber’s mode is described by the equations below.
This model requires a 0.3 eV energy gap between the donor and acceptor Lumos to achieve successful charge transfer, as shown in Equationeq 4(4)
(4) . However, this LUMO offset should not be confused with the empirical shifts of 0.3 eV for Equationeq 5
(5)
(5) . As a result, the highest value for eVOC is Eopt = 0.6 eV.
The LUMO level prediction is not as precise as the HOMO level prediction may potentially be a concern; nonetheless, DFT energy levels must be evaluated in the context of the resulting quantities of interest. According to Equationeqs 4(4)
(4) , Equation2
(2)
(2) , and Equation3
(3)
(3) , the LUMO level allows us to determine the optical band gap, LUMO offset, and open-circuit voltage. The optical band gap description is irrelevant in this scenario because the LUMO level description has been fitted to the bandgap. Because the LUMO offset criteria are required to enable a functional device, donor polymers with LUMO values close to the 0.3 eV limit of Equationeq 2
(2)
(2) may encounter difficulties. A higher level of uncertainty in the LUMO level value, on the other hand, is not an issue for values that are far from the offset limit.
In general, the optimization of polymer-fullerene solar cells is based on fine-tuning the electronic properties and interactions of the donor and acceptor components in order to absorb the most light and generate the most free charges while consuming the least amount of energy and transporting the charges to the electrodes. However, in order to develop next-generation, high-efficiency solar systems, it is vital to understand the optimal electronic characteristics that each component should have. For a variety of reasons, fullerenes are now regarded as the best acceptors for organic solar cells. For starters, they contain an energetically deep-lying LUMO, which gives the molecule an extremely high electron affinity in comparison to the numerous possible organic donors [Citation43].
Except for charge collection, which is based on the electrical contact between the active layer composite and the appropriate electrode, it is clear that the active layer donor-acceptor composite now regulates all aspects of the mechanism. The energy difference between the donor’s HOMO and the acceptor’s LUMO is shown to be extremely closely connected with the VOC value [Citation6,Citation44,Citation45]. Therefore, the maximal open-circuit voltage (VOC) of the BHJ solar cell is proportional to the difference between the electron donor’s highest occupied molecular orbital (HOMO) and the electron acceptor’s LUMO, after accounting for energy lost during photo charge creation [Citation46]. The following expression was used to compute the theoretical values of open-circuit voltage VOC:
All donors in this investigation were computed using the [6,6]-phenyl-C60-butyric acid methyl ester (PC60BM) fullerene derivative as the electron acceptor and the VOC values are shown in . These values are adequate for an efficient electron injection system. As a result, all compounds investigated can be employed as organic solar cell components since electron injection from the excited molecule to the acceptor’s conduction band (PC60BM), and subsequent regeneration is achievable in photovoltaic cells.
Table 3. The open circuit voltage VОС (eV) and First singlet excitation energy (Eoрt), exciton binding energy (EB) of the studied D-A monomers
3.4. Optoelectronic parameters
3.4.1. Electronic absorption spectra
The excitation energy based on the first and second singlet-singlet electronic transitions has been examined to understand the electronic characteristics of D-A-D monomers better. Time-dependent density functional theory (TD-DFT) has evolved as a trustworthy standard method for the theoretical study of electronic excitation spectra in recent years, with recent publications demonstrating improved accuracy for a wide range of systems [Citation47,Citation48]. Based on the improved geometry, the TD-DFT/B3LYP/6-311 G was employed. and show the nature and energy of the electronic transitions of all monomers in all series under investigation in the gas and solvent phases. All electronic changes are type, and there are no localized electronic transitions among the estimated singlet-singlet transitions. In addition, and summarize the absorption spectral transition energies and oscillator strength (f).
Table 4. The Calculated electronic transition data obtained using TD/DFT/B3LYР/6-311 G fоr аll D-А monomers in the gas
Table 5. The Calculated electronic transition data obtained using TD/DFT/B3LYР/6-311 G fоr аll D-А monomers in the chlorobenzene solvent
Approximately 70% of the solar photon flux is dispersed in the wavelength range of 380 (3.26 eV) to 900 nm (1.38 eV) [Citation6]. As a result, to maximize the number of excitons created and raise the Jsc, the D/A blend should have a broad and robust absorption in the same region. Because PC60BM (as an acceptor) exhibits poor absorption in this region of the solar spectrum (max = 470 nm), the donor molecule must serve as the primary light absorber. As a result, the photoexcitation properties of the 1–9 were investigated to understand better the physical mechanisms involved in photocurrent generation. In addition, the TDDFT method has been used to predict the electronic transition energies of p-conjugated molecules as a cost-effective method [Citation49].
and show that the excitation energy for all substances investigated is about 2 eV. Furthermore, all monomers had absorption maxima in the 539–739 nm wavelength range in gas and 586–758 nm range in the solvent phase, showing that all molecules only have one band in the visible region (abs > 400 nm). These compounds’ absorption spectra revealed a prominent absorption band attributed to the local transition electron. According to the transition nature, the HOMO LUMO transition is the first singlet excitation in the majority of the compounds. show the maximum (max) wavelengths for UV-vis absorption spectra of all chemicals simulated in the gas and chlorobenzene solvents.
The geometries of our proposed systems’ absorption spectra appear to be consistent with the usual model for other D-A molecules, which shows an absorption spectrum with a more excellent high-energy absorption band than a weaker low-energy absorption band [Citation50]. D-A-D molecules’ unique absorption spectrum differs from the one broad absorption peak observed in standard p-conjugated polymers such as P3HT [Citation51].
3.4.2. Effect of solvent
Because most chemical activities occur in the solution phase, solvent effects garner a lot of interest. Therefore, the solvent effects are included here to guarantee that the calculations are compatible with the typical conditions. The following section goes into greater detail about the impact of the chemical environment. Excitation energies and maximum absorption quantum chemical computations on the examined molecules were performed in vacuum, in chlorobenzene, to investigate the effect of solvents on the ground state molecular geometry. The orders of magnitude of the spectrum changes range from 9.3 nm to 64.3 nm 46.92 nm for all D-A-D monomers examined.
3.5. Density of states (DOS)
The DOS plot is a valuable tool for demonstrating molecular orbitals and their importance to chemical bonding. The results of the DOS plot demonstrate an overlapping population in the molecular orbital. The DOS plot shows the composition of the orbital group that contributes to the molecular orbital. depicts the DOS graphs in the gas phase. The density of localized states has a dramatically rising tendency to arise in the 7.5 to 13 eV range. The graph depicts the orbital properties of various energy ranges. It is the primary contribution of carbon’s s orbital and p essential orbital functions in the frontier molecular orbital. Because electron injection from the excited molecule to the conduction band of the acceptor (PC60BM) and subsequent regeneration is allowed in a photovoltaic cell, all of the compounds investigated can be employed as organic solar cell components based on VOC values.
4. Conclusions
The B3LYP hybrid functional with 6–31 g (d) basis set is used to theoretically investigate a donor-acceptor-donor (D-A-D) type 3,6-carbazole (CB) paired with different -conjugated benzothiazole derivatives monomer. We also estimated the energy difference between the HOMO and LUMO energies. The optical characteristics of polymers, on the other hand, were modelled using time-dependent DFT at the B3LYP level of theory. The HOMO level of the donor and the LUMO level of the acceptor molecule affect the open-circuit voltage of bulk-heterojunction devices utilizing PCBM as acceptor. When compared to other monomers, the lower Eg of CB-TDP-CB and CB-SDP-CB illustrates the importance of intramolecular charge movement, which causes the ingestion spectra to redshift. This is clearly due to the impact of the electron-contributor unit, which is more solid for 3,6-CB-TDP-CB and 3,6-CB-SDP-CB than for other monomers. All monomers had absorption maxima in the 539–739 nm range in gas and 586–758 nm range in the solvent phase, showing that all molecules contain only one band in the visible region (abs > 400 nm). The morphologies of the absorption spectra of our constructed systems appear to be equivalent to the usual model for other D-A molecules, indicating an absorption spectrum with a greater high-energy absorption band than a weaker low-energy absorption band.
Scheme 1. Building units as donor/acceptor moieties
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Wolf J, Babics M, Wang K, et al. Benzo[1,2-b: 4,5-b′]dithiophene-pyrido[3,4-b]pyrazine small-molecule donors for bulk heterojunction solar cells. Chem Mater. 2016;28(7):2058–2066
- Wang M, Cai D, Yin Z, et al. Asymmetric‐ indenothiophene‐based copolymers for bulk heterojunction solar cells with 9.14% efficiency. Adv Mater. 2016;28(17):3359–3365
- Scharber MC, Sariciftci NS. Efficiency of bulk-heterojunction organic solar cells. Prog Polym Sci. 2013;38(12):1929–1940
- Li G, Zhu R, Yang Y., et al. Polymer solar cells. Nat Photon. 2012;6(3):153–161
- Green MA, Emery K, Hishikawa Y, et al. Dunlop ED solar cell efficiency tables. 45th version. Prog. Photovoltaics Res. Appl. 2015; 23:1–9.
- Scharber MC, Mühlbacher D, Koppe M, et al. Design rules for donors in bulk‐heterojunction solar cells—towards 10% energy‐ conversion efficiency. Adv Mater. 2006;18(6):789–794
- Heeger AJ. Semiconducting and metallic polymers: the fourth generation of polymeric materials (nobel lecture). Angew Chem Int Ed Engl. 2001;40(14):2591–2611.
- Heeger AJ. Semiconducting and metallic polymers: the fourth generation of polymeric materials. J Phys Chem B. 2001;105(36):8475–8491.
- Facchetti A. π-conjugated polymers for organic electronics and photovoltaic cell applications. Chem Mater. 2011;23(3):733–758.
- Ullah H, Tahir AA, Mallick TK., et al. Polypyrrole/TiO2 composites for the application of photocatalysis. Sens Actuators B. 2017;241:1161–1169.
- Ullah H. Inter-molecular interaction in polypyrrole/TiO2:A DFT study. J Alloys Compd. 2017;692:140–148.
- Mishra A, Ma CQ, Bäuerle P., et al. Functional oligothiophenes: molecular design for multidimensional nanoarchitectures and their applications. Chem Rev. 2009;109(3):1141–1276
- Perepichka IF, Perepichka DF. Handbook of Thiophene-Based Materials: applications in Organic Electronics and Photonics, 2 Volume Set. Chichester: John Wiley & Sons; 2009
- Li M, An C, Pisula W, et al. Cyclopentadithiophene−benzothiadiazole donor−acceptor polymers as prototypical semiconductors for high-performance field-effect transistors. Acc Chem Res. 2018;51(5):1196–1205.
- Farinola GM, Ragni R. Electroluminescent materials for white organic light emitting diodes. Chem Soc Rev. 2011;40(7):3467–3482.
- Hacioglu SO, Toksabay S, Sendur M, et al. Synthesis and electrochromic properties of triphenylamine containing copolymers: effect of π-bridge on electrochemical properties. J Polym Sci Part A: Polym Chem. 2014;52(4):537–544.
- Bhargav R, Bhardwaj D, Shahjad PA, et al. Poly(Styrene Sulfonate) free Poly(3,4-Ethylenedioxythiophene) as a robust and solution-processable hole transport layer for organic solar cells. ChemistrySelect. 2016;1(7):1347–1352.
- Zarubin VA, Li TD, Humagain S, et al. Improved anisotropic thermoelectric behavior of poly(3,4-ethylenedioxythiophene): poly(styrenesulfonate) via magnetophoresis. ACS Omega. 2018;3(10):12554–12561.
- Echeverri M, Martín I, Concellon A, et al. Noise and its effect on the health of personnel in the dental section of the CES specialist center.CES Odontol. 1989; 2(1):13–20. Gutierrez-Puebla, E.; Serrano, J. L.; Go ́ mez-Lor, B. Fluorescent and Electroactive Monoalkyl BTD-Based Liquid Crystals with Tunable Self-Assembling and Electronic Properties. ACS Omega 2018, 3, 11857−11864.
- Chiu CC, Sheng YC, Lin WJ, et al. Effects of internal electron-withdrawing moieties in D−A− π−A organic sensitizers on photophysical properties for DSSCs: a computational study. ACS Omega. 2018;3(1):433–445.
- Li T, Dai S, Ke Z, et al. Fused Tris(thienothiophene)-based electron acceptor with strong near-infrared absorption for high-performance as-cast solar cells. Adv Mater. 2018;30(10):1-7. [PubMed: 29334151].
- Li P, Wang Z, Song C, et al. Rigid fused π-spacers in D–π–A type molecules for dye-sensitized solar cells: a computational investigation. J Mater Chem C. 2017;5(44):11454–11465.
- Hosseinzadeh E, Hadipour NL. The influence of the structural variations of the fused electron rich-electron deficient unit in the π-spacer of A-D-π-D-A organic dyes on the efficiency of dye-sensitized solar cells: a computational study. Org Electron. 2018;62:43–55.
- Boudreault P-LT, Beaupré S, Leclerc M., et al. Polycarbazoles for plastic electronics. Polym Chem. 2010;1(2):127–136.
- Boudreault PLT, Blouin N, Leclerc M., et al. Poly(2,7-carbazole)s and related polymers. In: Advances in Polymer Science. Berlin, Heidelberg, Germany: Springer; 2008. p. 99–124.
- Blouin N, Michaud A, Gendron D, et al. Toward a rational design of poly(2,7-carbazole) derivatives for solar cells. J Am Chem Soc. 2008;130(2):732–742.
- Wakim S, Aïch B, Tao Y, et al. Charge transport, photovoltaic, and thermoelectric properties of poly(2,7-carbazole) and poly(indolo[3,2-b]carbazole) derivatives. Polym Rev. 2008;48(3):432–462.
- Nandy BC, Gupta AK, Mittal A, et al. Carbazole: it’s biological activity. J. Biomed. Pharm Res. 2014;3:42–48.
- Ö Y, Sezer E, Saraç AS., et al. Spectroelectrochemical study of N-ethyl-carbazole in the presence of acrylamide. Polym Int. 2001;50(3):271–276.
- Ghosh S, Bedi A, Zade SS., et al. Thienopyrrole and selenophenopyrrole donor fused with benzotriazole acceptor: microwave assisted synthesis and electrochemical polymerization. RSC Adv. 2015;5(7):5312–5320.
- Vuai SAH, Babu NS. Theoretical design of low bandgap donor–acceptor (D-A) monomers for polymer solar cells: DFT and TD-DFT study. Des Monomers Polym. 2021;24(1):123–135.
- Babu NS, Vuai SAH. Theoretical studies of optoelectronic and photovoltaic properties of D–A polymer monomers by density functional theory (DFT). Des Monomers Polym. 2021;24(1):224–237.
- Mohr T, Aroulmoji V, Ravindran RS, et al. DFT and TD-DFT study on geometries, electronic structures and electronic absorption of some metal free dye sensitizers for dye sensitized solar cells. Spectrochim Acta A Mol Biomol Spectrosc. 2015;135:1066–1073.
- Xie XH, Shen W, He RX, et al. A density functional study of furofuran polymers as potential materials for polymer solar cells. Bull Korean Chem Soc. 2013;34(10):2995–3004.
- Becke AD. Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys. 1993;98(7):5648–5652.
- Frisch MJ, Trucks GW, Schlegel HB., et al. Gaussian 09. Revision B.03. Pittsburgh, PA: Gaussian, Inc; 2009.
- Parr G, Yang W. Density-Functional Theory of Atoms and Molecules. Oxford, NY: University Press; 1989.
- Tomasi J, Mennucci B, Cammi R., et al. Quantum mechanical continuum solvation models. Chem Rev. 2005;105(8):2999–3093.
- Kurashige Y, Nakajima T, Kurashige S, et al. Theoretical investigation of the excited states of coumarin dyes for dye-sensitized solar cells. J Phys Chem A. 2007;111(25):5544–5548.
- Zhou H, Yang L, You W., et al. Rational design of high performance conjugated polymers for organic solar cells. J Mater Chem C. 2013;1(44):7266–7293. Macromolecules. 2012;45(2):607-632; b) Wang T, Pearson AJ, Lidzey DG. Correlating molecular morphology with optoelectronic function in solar cells based on low band-gap copolymer:fullerene blends.
- Air mass ASTM G-173 data available at. Accessed 2013 Apr 9. http://rredc.nrel.gov/solar/spectra/am1.5/
- Brédas JL, Beljonne D, Coropceanu V, et al. Charge- ́ transfer and energy-transfer processes in π-conjugated oligomers and polymers: a molecular picture. Chem Rev. 2004;104(11):4971–5004.
- Halls JJM, Cornil J, dos Santos DA, et al. Charge- and energy-transfer processes at polymer/polymer interfaces: a joint experimental and theoretical study. Phys Rev B. 1999;60(8):5721–5727.
- Gunes S, Neugebauer H, Sariciftci NS., et al. Conjugated ¨ polymer-based organic solar cells. Chem Rev. 2007;107(4):1324–1338.
- Brabec CJ, Cravino A, Meer D, et al. Origin of the open circuit voltage of plastic solar cells. Adv Funct Mater. 2001;11(5):374–380.
- Gadisa A, Svensson M, Andersson MR, et al. Correlation between oxidation potential and open-circuit voltage of composite solar cells based on blends of polythiophenes/fullerene derivative. Appl Phys Lett. 2004;84(9):1609–1611.
- Kaneto K, Yoshino K, Inuishi Y., et al. Electrical and optical properties of polythiophene prepared by electrochemical polymerization. Solid State Commun. 1983;46(5):389–391.
- Jacquemin D, Preat J, Wathelet V, et al. Thioindigo dyes: highly accurate visible spectra with “TD-DFT”. J Am Chem Soc. 2006;128(6):2072–2083.
- Adamo C, Jacquemin D. The calculations of excited-state properties with time-dependent density functional theory. Acc Chem Res. 2012;45(8):1173–1182. Chem Soc Rev. 2013;42(3):845-856; b) Perrier A, Maurel F, Jacquemin D. Single molecule multiphotochromism with diarylethenes.
- Kanibolotsky AL, Vilela F, Forgie JC, et al. Well-defined and monodisperse linear and star-shaped quaterfluorene-DPP molecules: the significance of conjugation and dimensionality. Adv Mater. 2011;23(18):2093–2097.
- Khlaifia D, Ewels CP, Massuyeau F, et al. Unraveling the real structures of solution-based and surface-bound poly(3-hexylthiophene) (P3HT) oligomers: a combined theoretical and experimental study. RSC Adv. 2016;6(61):56174–56182.