Abstract
This paper presents the directly obtained rotational viscosity values of E7, which includes pentylcyanobiphenol, heptylcyanobiphenol, 4-cyano-4′ -n-octyloxy-1,1′ -biphenyl, and 4-cyano-4′′-n-pentyl-1,1′,1′′-terphenyl, at various temperatures using molecular dynamics computer simulation. The director mean squared displacement was achieved from the squared displacement of the mean director using the concept of the mean director of various nematic liquid crystals. The calculated values were compared with the experiment results that predicted a good agreement. Additional points that must be considered for further study are also discussed.
1. Introduction
It is well known that rotational viscosity is directly related to the response time of a liquid crystal (LC) display. The calculation of the rotational viscosity of LCs is one of the most challenging works in molecular dynamics (MD) computer simulation Citation1–9. Sufficient time is generally highly needed to achieve reliability in equilibrium MD simulation studies, but in the test in this study, it was found that to complete 5.0 million steps, an essential amount of time is enough. In this study, such technique was used to find the rotational viscosity of a mixture, E7, which included pentylcyanobiphenol (5CB), heptylcyanobiphenol (7CB), 4-cyano-4′ -n-octyloxy-1,1′ -biphenyl (8OCB), and 4-cyano-4″-n-pentyl-1,1′,1″-terphenyl (5CT).
The rotational viscosity of a mixture of 5CB and decylcyanobiphenol (10CB) was calculated in 2009 Citation9–13. It was the first time the rotational viscosity of a mixture of different kinds of LCs was calculated Citation10 Citation13.
Since the commercial LC mixture includes 15–20 different kinds of LCs, it is essential to add more kinds of LCs to the ensemble for the MD computer simulation.
In this paper, a method of directly calculating the rotational viscosity of the E7 mixture of four different kinds of LCs is presented as follows: 5CB, 7CB, 8OCB, and 5CT Citation14.
The overall methodology comprises molecular geometry optimization and charge calculation using the Gaussian ab initio calculation, the MD computer simulation of an LC mixture, and numerical analysis Citation10
Citation13. Some parameters of the Gaussian ab initio calculation were modified. As the current system of the simulation is more complicated than the earlier system (), the MD time of the equilibrium was first extended to lower the fluctuations of the mixture system (). The mean director vector data of the ensemble were used to calculate the mean squared displacement Citation10–13. This technique made it easy to perform numerical analysis after obtaining the data from the MD simulation. The rotational viscosity equation was derived from two equations: the Einstein relation and the Stokes–Debye–Einstein equation Citation7,Citation10–13. The essential factors of the rotational viscosity equation were the squared displacement of the mean director as a function of time and the mean volume and the mean temperature, which were obtained from the MD computer simulation. The average results were found, and better numerical analysis values were obtained. Eventually, the rotational viscosity of E7 can be calculated directly from each molecular structure and composition (). The direct calculation method makes it possible to predict the rotational viscosity even if only the molecular structure and the molar ratio are known. The research and development scientist or engineer can rapidly find the target mixture that has a lower rotational viscosity composition and molecular structure using this method in this industrial area. Moreover, the authors show their current work and discuss important points for further study.
Figure 1. Chemical molecular structures of 5CB and 10CB.
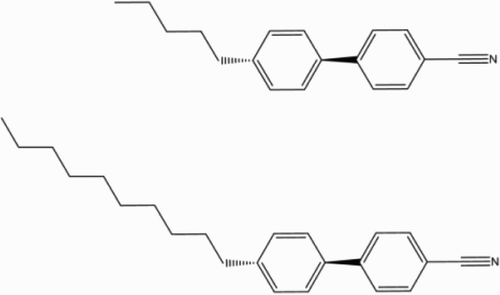
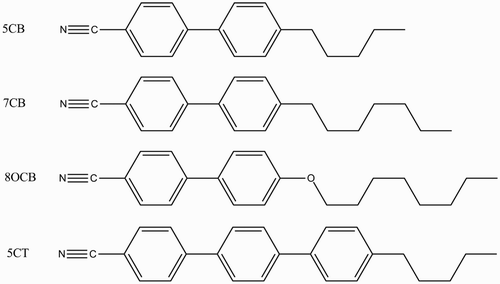
Figure 2. Chemical molecular structures of the E7 components.
2. Computation details
2.1. A sample
The sample MD computer simulation in this study consisted of the nematic LC mixture E7 and its components (shown in ) – 5CB, 7CB, 8OCB, and 5CT. The composition (w/w) of these four different kinds of LCs was as follows: 51% 5CB, 25% 7CB, 16% 8OCB, and 8% 5CT.
2.2. Overall workflow
In this study, the input parameters and MD simulation time parameters were modified. These steps were performed mainly to attain more accurate charge density values. The details of such studies can be seen in Refs. Citation11 Citation12. The whole process of the MD computer simulation is as follows ().
Figure 3. Overall MD workflow.
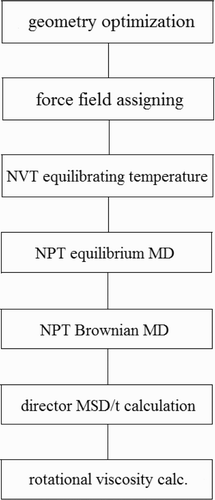
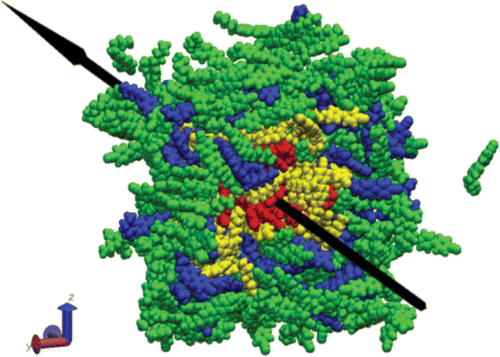
Figure 4. MD snapshot of the E7 mixture (the black arrow represents the nematic ‘mean director’).
As described, the modified input parameters of the ab initio calculation were utilized. The ab initio command parameters of the Gaussian package for the geometry optimization and the charge calculation are as follows, respectively:
For the optimization process, the keyword ‘opt’ was required. The geometry optimization and charge calculation were performed sequentially and separately. The molecules of the E7 mixture were modeled using Xleap, a program within the AMBER package. The atom type and charge distribution information was transferred from the result of the ab initio calculation to the MD model using Antechamber, another computer program within the AMBER package.
The generalized AMBER force field parameter, which is applicable to general organic molecules such as LCs and to the AMBER force field equation that follows, was assigned.
The force field for the bonded atoms consisted of bond energy, angle energy, and dihedral energy. The force field for the non-bonded molecules consisted of electrostatic energy and van der Waals energy as follows:
Table 1. Comparison of the MD times of 5CB+10CB and E7.
The input commands in Steps 2 and 3 are exactly the same, except for the MD run time. It was divided into two steps to investigate the sufficient equilibrium from the fluctuation during the MD computer simulation. Then Brownian dynamics followed for 2 ns.
The purpose of this step was to implement the natural state of an E7 nematic mixture composed of four different kinds of LCs. A very weak heat bath was used, however, to overcome the energy leakage phenomenon during the fourth MD. Eventually, the results were acquired by averaging the calculated figures many times to overcome numerical fluctuation. It was recently realized that this problem can be solved, and such conditions are generally being studied. Although MD runs in a parallel environment using openMPI and numerical analysis written in C, the code was optimized using openMP, and it took about 15 days to perform the overall computer simulation using a quad core CPU environment Citation15–17.
3. Results
3.1. Mean director of the mixture of four different kinds of LC molecules
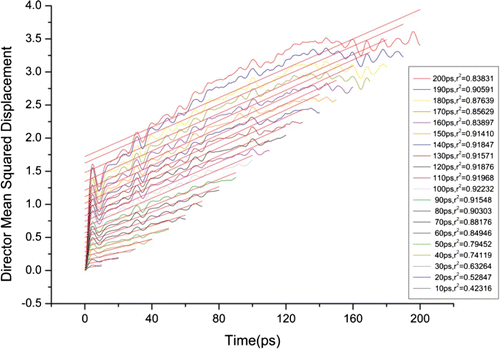
shows the E7 ensemble that included 5CB, 10CB, 8OCB, and 5CT. Nematic LCs have a dipole because they have many atoms and their electronegativities are different. Thus, they each have their own dipole moment. According to the continuum theory, nematic LCs could align themselves along the long axis. Eventually, the dipole moment could be the vector summed up over the long distance. Such dipole moment is called the ‘mean director,’ which is used to calculate the mean squared displacement. The mean squared displacement can be achieved only by calculating the squared displacement of the mean director vector. It has an advantage during the numerical analysis because of its ease and the lower numerical fluctuations over the averaging processes Citation10–13. shows the mean squared displacement with time, which was directly obtained from the squared displacement of the mean director. The mean director vector was read from the trajectory file after MD by ‘ptraj’, a program in the AMBER package.
Figure 5. Squared displacement of the mean director of E7.
3.2. Rotational viscosities
The rotational viscosity equation was derived from the Stokes–Einstein–Debye equation and the Einstein relation, as follows:
compares the rotational viscosities of the calculated and experimental values at various temperatures.
Table 2. Comparison of the rotational viscosities of the calculated and experimental values at various temperatures.
3.3. Recommendations for further study
The points that must be considered for further study are as follows.
-
The generalized AMBER force field is highly recommended for organic molecules such as LCs Citation18.
-
A parallel computing environment, especially the symmetry multi-processor type, is recommended for low latency instead of the cluster computer system using a 100 MB LAN.
-
T he ensemble system should be equilibrated sufficiently in the equilibrium MD simulation work.
-
T he squared displacement of the mean director vector is recommended to calculate the mean squared displacement [10–13 .
-
T he determination coefficients of the fitted lines of the squared displacement of the mean director with time can be used to select better calculated rotational viscosity values. Later, the selected values should be averaged from the various lengths of the mean squared displacement of the mean director [10–13 .
-
T he calculated values can be obtained through longer-time computer calculation. Using openMP or heterogeneous openCL, computing would be helpful in the numerical calculation, because such packages can be used with all CPU and
systems, respectively [15–17 .
4. Conclusion
The rotational viscosity of the E7 mixture was calculated for temperatures that ranged from 10°C to 40°C, using a more complicated system than that used in the authors’ previous study () Citation10
Citation13. The investigation was performed using the concept of the mean director of the LC mixture, which included four different kinds of LCs. A longer equilibrium MD time was experimentally used to equilibrate the E7 system. The authors are currently studying an interdisciplinary area related to this study.
Acknowledgements
The authors acknowledge Yong-Kuk Yun, Ph.D., and Seung-Eun Lee, Ph.D., from Merck Korea, for providing the experimental rotational viscosity data on the E7 mixture; Sang Soo Han, Ph.D., from the Korea Research Institute of Standards and Science for the detailed discussions; Kwang Jin Oh, Ph.D., and Ji-Hoon Jang from the Supercomputing Center of the Korea Institute of Science and Technology Information for the detailed discussions and for their support with the partial computing resources; Junard Lee and Hyungon Ryu from NVIDIA and David Yang from Miruware for their partial support with the computing resources; and Mr. Min Cheol Han from the Department of Nuclear Engineering of Hanyang University for his discussion on openMP.
References
- de Gennes , P. G. and Porst , J. 1994 . The Physics of Liquid Crystals , Oxford, , UK : Clarendon Press .
- Smondyrev , M. , Loriot , G. B. and Pelcovits , R. A. 1995 . Phys. Rev. Lett , 75 : 2340
- Sarman , S. 1997 . Phys. A , 240 : 160
- Demus , D. , Goodby , J. , Gray , G. W. , Spiess , H. W. and Vill , V. 1998 . Handbook of Liquid Crystals , Weinheim, , Germany : Wiley-VCH Press . Vol. 2A
- Zakharov , A. V. , Komolkin , A. V. and Maliniak , A. 1999 . Phys. Rev. E , 59 ( 6 ) : 6802
- Kuwajima , S. and Manabe , A. 2000 . Chem. Phys. Lett , 332 : 105
- Cheung , D. L. , Clark , S. J. and Wilson , M. R. 2002 . Chem. Phys. Lett , 356 : 140
- Gupta , S. D. and Roy , S. K. 2003 . Phys. Lett. A , 306 : 235
- Ilk Capar , M. and Cebe , E. 2005 . Chem. Phys. Lett , 407 : 454
- Kim , J.-S. , Ahmad , F. , Muhammad , J. , Park , S.-W. , Lee , J.-W. , Yun , H.-Y. , Jung , J. E. , Jang , J. E. , Jeon , Y.-J. and Kim , Y.-B. 2009 . IMID 2009 Tech. Dig , : 607
- Kim , J.-S. , Jamil , M. , Jung , J. E. , Jang , J. E. , Ahmad , F. , Lee , J. W. , Park , S. W. and Jeon , Y. J. 2010 . IMID/IDMC/Asia Display 2010 Tech. Dig , 664
- Kim , J.-S. , Jamil , M. , Jung , J. E. , Jang , J. E. , Lee , J. W. , Ahmad , F. , Woo , M.-K. , Kwak , J. Y. and Jeon , Y. J. presented at the Korea Liquid Crystal Conference . pp. 58 – 61 . Yeungnam University .
- Kim , J.-S. , Jamil , M. , Jung , J. E. , Jang , J. E. , Ahmad , F. , Lee , J. W. , Park , S. W. , Woo , M.-K. , Kwak , J. Y. and Jeon , Y.-J. 2011 . Chin. J. Chem , 29 : 48
- Brás , A. R.E. 2005 . Electronic – Liquid Crystal Communications 1; A publication of the International Liquid Crystal Society, http://www.e-k.org
- Open MPI: Open Source High Performance Computing, http://www.open-mpi.org/
- The OpenMP API Specification for Parallel Programming, http://openmp.org/wp/
- OpenCL is the open standard for parallel programming of heterogeneous systems, http://www.khronos.org/opencl/
- (a) J. Wang, R.M. Wolf, J.W. Caldwell, P.A. Kollman, D.A. Case, Comput. Chem. 26, 114 (2005); (b) http://ambermd.org.