
Abstract
Total suspended particle (TSP) samples were collected at Cape Hedo, Okinawa on a basis of 24, 12 and 3 h intervals in March to April 2007, when the Asian outflow is enhanced. The filter samples were analysed for dicarboxylic acids, oxoacids, benzoic acid and α-dicarbonyls to better understand the sources, chemical composition and photochemical ageing of organic aerosols during long-range transport. Their molecular distributions showed a predominance of oxalic acid (C2) followed by malonic (C3) and succinic (C4) acids. Day/night samples did not show clear diurnal variations, suggesting that local anthropogenic influence is not important. The highest concentrations of C2 and other diacid species were observed in April 10 when air masses were delivered from north China. Higher concentrations were observed when air masses arrived from East Asia and coastal China. The 3 h samples showed higher concentrations of oxalic acid at 16:30 or at 13:30 during April 14 and 16, whereas higher relative abundance of C2 in total diacids and higher concentration ratios (C2/C3, C2/C4, C3/C4, C2/Gly, and C2/ωC2) were observed at 7:30 during April 14–16. These results suggest that aerosols were accumulated in the atmosphere during night and photochemically modified early in the morning. Oxalic acid–C/OC did not show a prominent peak in 3-h samples. Temporal variations in the concentrations of diacids, oxoacids and α-dicarbonyls, relative abundances and diacid–C/OC ratios together with 5 day back trajectory analyses show that the aerosols from Cape Hedo are strongly influenced by long-range atmospheric transport from East Asia, especially in China. Strong correlations of C2 with nss-SO42− and NO3− in night-time suggest a secondary formation of water-soluble diacids via heterogeneous aqueous phase oxidation during long-range transport.
1. Introduction
Atmospheric aerosols contain high abundance of water-soluble dicarboxylic acids (Grosjean et al., Citation1978). The abundant presence of diacids in aerosols increases their hygroscopic properties (Saxena et al., Citation1995), affecting their cloud condensation nuclei and ice nuclei activities (Novakov and Penner, Citation1993). Small organic acids may also have an adverse effect on human health (Highwood and Kinnersley, Citation2006). Chemical analysis of ambient aerosols from Okinawa, an outflow region of Asian dusts (Kunwar and Kawamura, Citation2014a) and transmission electron microscopy analysis of soot particles from Okinawa (Ueda et al., Citation2011) showed that aerosols are significantly aged during long-range atmospheric transport and heavily influenced from East Asian outflow. There are various sources of diacids in East Asia, including fossil fuel combustion (Ho et al., Citation2006), biomass burning of agricultural wastes (Kawamura et al., Citation2013), meat cooking operations (Fu et al., Citation2013) and vehicular emission (Wang et al., Citation2010) as well as secondary photo-induced oxidations of biogenic unsaturated fatty acids (Kawamura and Gagosian, Citation1987; Pokhrel et al., Citation2015, Citation2016). However, the secondary production of diacids overwhelms the primary emission in the atmosphere (Kawamura, Steinberg et al., Citation1985; Kawamura et al., Citation2010; Kawamura and Bikkina, Citation2016).
Cape Hedo of Okinawa Island is situated in the outflow region of East Asia (Kunwar and Kawamura, Citation2014a) and has been used as a supersite of Atmospheric Brown Clouds (ABC) project to study the atmospheric transport of Chinese aerosols and pollutants and their chemical transformation (Takami et al., Citation2007). The consumption and emission of anthropogenic sources are increasing day by day in East Asia (Ohara et al., Citation2007). Previously, we reported one-year observation on molecular distributions of diacids and related compounds and carbonaceous components in the Okinawa aerosols to better understand the seasonal variations of those water-soluble organics in the western North Pacific Rim (Kunwar and Kawamura, Citation2014a, 2014b). Although there are few studies of diacids and related compounds in Okinawa and its surroundings (Kunwar and Kawamura, Citation2014a; Kundu et al., Citation2010; Deshmukh et al., Citation2016), there is no time series (e.g. 12 and 3 h) study to decipher the source and photochemical formation of organic acids. The lack of such studies motivates us for collecting time-resolved samples in this region. We identified a time-specific photochemical process of diacids and related compounds, which provides very useful information for future aerosol studies.
In this study, we report molecular distributions of diacids, oxoacids, pyruvic acid, benzoic acid and α-dicarbonyls in ambient aerosols from Cape Hedo, Okinawa on a basis of daily (24 h) and diurnal intervals (day/night and 3 h) to better understand the sources of organic aerosols and in situ photochemical production of water-soluble organic species during long-range atmospheric transport. Based on principal component analysis (PCA), back trajectory analysis, correlation analysis with some specific tracers, we discuss the sources and compositional changes of water-soluble organics in the time-resolved samples, and the photochemical aging of organic aerosols.
2. Samples and analytical procedures
2.1. Site description
Figure shows the sampling site of Cape Hedo, Okinawa Island. Aerosol samples were collected at Cape Hedo Atmosphere and Aerosol Monitoring Station (CHAAMS, 26º 9′ N, 128° 2′ E), located on the north-west coast of subtropical Okinawa Island. There are no major anthropogenic activities in Cape Hedo, which is surrounded by subtropical rain forest (Takami et al., Citation2007; Sato et al., Citation2009; Yamamoto and Kawamura, Citation2011; Kunwar, Kawamura, et al., Citation2016; Kunwar, Torii, et al., Citation2016). In the Asian Pacific region, the East Asian monsoon dominates in spring, delivering continental air masses from East Asian countries over Okinawa (Hatakeyama et al., Citation2001; Kunwar and Kawamura, Citation2014a, 2014b).
Fig. 1. A map of East Asia with sampling location of Cape Hedo, Okinawa, Japan.
2.2. Aerosol sampling
Total suspended particles (TSP) were collected on a basis of 24 h for 21 days (March 15–26 and April 2–10), 12 h for 9 days (March 27 to April 1 and April 11–13) and 3 h for 4 days (April 14–17) in CHAAMS station during the campaign from March 15 to April 17, 2007. Aerosol samples (n = 55) were collected using a high-volume air sampler (Kimoto AS-810B) and pre-combusted (450 °C, 4 h) quartz fibre filters (Pallflex 2500QAT-UP, 20 × 25 cm). Before and after the sampling, filters were preserved in a pre-combusted glass jar (150 mL) with a Teflon-lined screw cap. The sample filters were stored in darkness at −20 °C until analysis. Field blanks were also collected from the site. For the data presentation, we have taken centre time of sampling period. It should be noted that the sample artefact might occur due to evaporation/adsorption process of semi-volatile organics on the filter or particle surface during sampling.
2.3. Chemical analysis
Low molecular weight diacids, oxoacids, and α-dicarbonyls were analysed using methods described in Kawamura et al. (Citation1996). Briefly, known area of filter was extracted with organic-free ultrapure water under ultra-sonication for 10 min (10 mL × 3). The extracts were passed through a glass column (Pasteur pipette) packed with quartz wool to remove insoluble particles and filter debris, and then concentrated near to dryness using a rotary evaporator under vacuum. Carboxyl and aldehyde groups were then derivatized to dibutyl esters and ω,ω-dibutoxy acetals, respectively, using 14% boron trifluoride (BF3)/n-butanol at 100 °C for 1 h, whereas keto group of pyruvic acid was converted to dibutoxy acetal. The derivatives were dissolved in n-hexane and quantified using a capillary gas chromatograph (GC; HP 6890). Identifications of the compounds were performed by comparing GC retention times with those of authentic standards and confirmed by mass spectral examination using GC/MS technique (Kawamura and Ikushima, Citation1993; Kawamura and Bikkina, Citation2016).
Recovery and reproducibility tests were performed before real sample analysis. The recovery for oxalic acid (C2) was 85%, more than 90% for C3, C4, C5 and C6 (Kunwar and Kawamura, Citation2014b) and 90% for glyoxal (Kunwar and Kawamura, Citation2014b). Kawamura and Yasui (Citation2005) reported that recovery of ωC2 is 88%. The analytical errors in the triplicate analysis were less than 5% for C2, C3, C4, C5 and C6. We also analysed field blanks. Concentrations of all the species reported here are corrected for the field blanks, which are <5% of real samples.
Organic carbon (OC) and elemental carbon (EC) were measured using a Sunset Laboratory carbon analyser following the IMPROVE temperature protocol (Wang et al., Citation2005). The method produced data for four OC fractions (OC1, OC2, OC3 and OC4 in a helium atmosphere at 140, 280, 480 and 580 °C, respectively), a pyrolyzed carbon fraction (OP, determined when reflected laser light attained its original intensity after oxygen was added to the combustion atmosphere), and three EC fractions (EC1, EC2 and EC3 in a 2% oxygen/98% helium atmosphere at 580, 740 and 840 °C, respectively). The IMPROVE protocol defined OC as OC1 + OC2 + OC3 + OC4 + OP and EC as EC1 + EC2 + EC3 − OP. The analytical errors in duplicate analysis of the filter sample were less than 8% for OC. OC concentrations reported here are corrected for the field blanks, which are less than 10% of real samples.
2.4. Backward air mass trajectory analysis
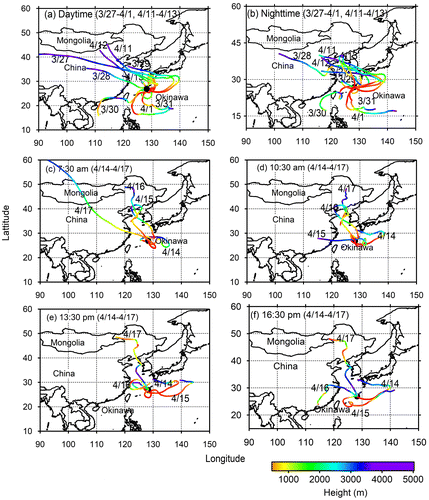
Five-day backward trajectory analyses were performed at 500 metres above the ground with height for 24, 12 and 3 h samples to decipher the source regions of air masses over Cape Hedo, using the Hybrid Single-Particle Lagrangian Integrated Trajectory (HYSPLIT4) model (https://www.arl.noaa.gov/ready/hysplit4.html), NOAA Air Resources Laboratory, Silver Spring, Maryland, United States (Draxler and Hess, Citation1998). Figure shows the air mass trajectories calculated for day and night samples and every 3 h samples. During March 30 (day and night) air masses travel the coastal area of south China, Taiwan and the Pacific. In contrast, during March 23, 24, 31 and April 1 air masses came from the Pacific Ocean. Except for the above-mentioned dates, all the air masses originated from East Asia. For each sample, we have calculated one back trajectory. We performed back trajectory (BT) analysis using geopotential height, time and model vertical velocity. Sampling collection time is the starting time for BT analysis.
Fig. 2. Plots of 5-day backward air mass trajectories (BT) (a) daytime, (b) night-time, (c) early morning, (d) late morning, (e) early afternoon and (f) late afternoon arriving in Cape Hedo at 500 m above the ground level (AGL) during the study periods.
2.5. Principal component analysis
Principal component analysis (PCA) is a mathematical algorithm that reduces dimensionality of the data while variation is mostly retaining in the data-set. It accomplishes reduction by identifying directions, called principal components, along which the variation in the data is maximal. Using a few components, each sample can be represented by relatively few groups instead of thousands of variables. PCA is a useful statistical tool for data analysis to assess the sources of aerosols. This method has been applied especially when sources of aerosols are different (Hopke, Citation1985; Kawamura and Sakaguchi, Citation1999). In this study, the data-sets of diacids from Okinawa (n = 55) were subjected to PCA analysis to investigate their sources and possible formation pathways. The term ‘loading’ in PCA represents the correlation coefficient between the component and individual species.
3. Results and discussion
3.1. Molecular distributions of dicarboxylic acids, oxoacids and α-dicarbonyls
We detected a homologous series of α,ω-dicarboxylic acids (C2–C12), ω-oxocarboxylic acids (ωC2–ωC9), pyruvic acid, α-dicarbonyls (glyoxal and methylglyoxal) and benzoic acid in the aerosol samples from Okinawa. Oxalic acid (C2) was found as the most abundant species followed by malonic (C3) and succinic (C4) acid (see Fig. ), which is consistent with previous studies of coastal marine aerosols (Kundu et al., Citation2010, Kunwar and Kawamura Citation2014b) and remote marine aerosols from the western Pacific (Sempéré and Kawamura, Citation2003). Among oxoacids, ωC2 is most abundant. Such molecular characteristics have been reported in previous studies of ambient aerosols from East Asia (Kawamura and Yasui, Citation2005; Kundu et al., Citation2010).
Fig. 3. Temporal variations in the concentration of (a) Total diacids (b) oxalic acid (C2), (c) malonic acid (C3), (d) succinic acid (C4), (e) adipic acid (C6), (f) pimelic acid (C7), (g) azelaic acid (C9), (h) phthalic acid, (i) terephthalic acid (tPh), (j) maleic acid (M), and (k) fumaric acid (F) in the ambient aerosols from Cape Hedo, Okinawa in spring 2007.
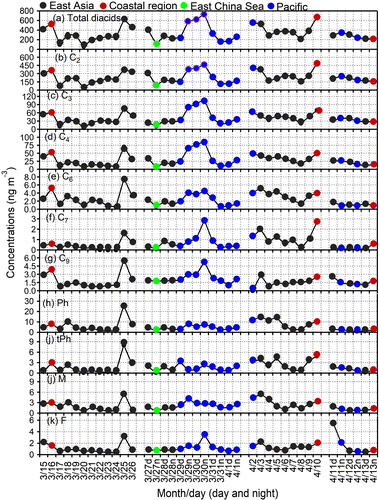
Table summarizes concentrations of diacids, oxoacids, pyruvic acid and α-dicarbonyls detected in the Cape Hedo aerosols together with their abbreviations. Concentrations of total diacids, total oxoacids and α-dicarbonyls during whole campaign ranged from 26 to 671 ng m−3 (av. 345 ± 150 ng m−3), 1.6 to 111 ng m−3 (30 ± 23 ng m−3), 1.3 to 58 ng m−3 (8.9 ± 9.8 ng m−3), respectively. The average concentrations of total diacids (345 ng m−3), oxoacids (30 ngm−3) and α-dicarbonyls (8.9 ng m−3) in Okinawa aerosols are lower than those (760, 45 and 9 ng m−3, respectively) reported in Beijing during August/September (Ho et al., Citation2010), and those (660, 53 and 12 ng m−3, respectively) from Gosan site, Jeju Island, an outflow region of Chinese aerosols (Kundu et al., Citation2010). Their concentrations are several times lower than those (1702, 242, 68 ng m−3, respectively) reported from the North China Plain (Kawamura et al., Citation2013). Further, concentrations of total diacids in springtime Okinawa aerosols are lower than those reported in the East China Sea (850 ng m−3) and Sea of Japan (1200 ng m−3) (Mochida et al., Citation2003).
Table 1. Concentrations of diacids, oxoacids, benzoic acid and α-dicarbonyls in the TSP samples collected from Okinawa in spring, 2007.
However, the concentrations of Okinawa aerosols are 1.7 times higher than those (200 ng m−3) from the western North Pacific (>140°E) (Mochida et al., Citation2003) and 8 times higher than remote marine aerosols (40 ng m−3) from the central Pacific (Kawamura and Sakaguchi, Citation1999). These comparisons suggest that Okinawa aerosols are significantly enriched with diacids, oxoacids and α-dicarbonyls compared to remote marine aerosols, but less enriched compared to the East Asian aerosols in source regions.
Figure S1 shows the molecular distribution of diacids, oxoacids and α-dicarbonyls during day and night-time samples. C2 is the dominant diacid species followed by C3, C4 and ωC2 in daytime, whereas benzoic acid is the dominant species after C4 during night-time. The higher concentration of benzoic acid in night-time suggests more influence from anthropogenic activities via long-range atmospheric transport. Indeed, benzoic acid has been identified as a direct pollutant from the traffic emissions (Kawamura, Ng et al., Citation1985) and indirect pollutant produced from photo-degradation of aromatic compounds (e.g. toluene) released from traffic exhausts (Suh et al., Citation2003). Duan et al. (Citation2008) suggested that oxidation of toluene is one of the significant sources of benzoic acid in the air. As described in experimental section, local anthropogenic activities are not important, thus, abundant presence of aromatic monoacids in the study site suggests a significant influence from anthropogenic activities related to traffic emissions in the source regions via long-range atmospheric transport.
3.2. Temporal variation of diacids, oxoacids and α-dicarbonyls in springtime Okinawa aerosols
Figure shows the temporal (daily and day/night) variations in the concentrations of selected diacids, whereas Fig. presents those of oxoacids and α-dicarbonyls. The highest concentration of total diacids was observed at night on March 30 when air masses were originated from the Pacific Ocean and travelled over Taiwan before reaching to Okinawa. Higher concentrations were also observed during March 16, 25, 29 (night-time) and April 10. Similar peaks were observed for oxalic, malonic and succinic acids (Fig. b–d). Concentration trends of total oxoacids and α-dicarbonyls are similar to those of total diacids, suggesting a similar source or similar formation mechanism. Based upon the backward trajectories, four source regions of diacids and related compounds were found as (a) East Asia, (b) coastal region, (c) the East China Sea and (d) the Pacific Ocean. We found higher concentrations of those compounds when air masses arrived from coastal region of China and East Asia.
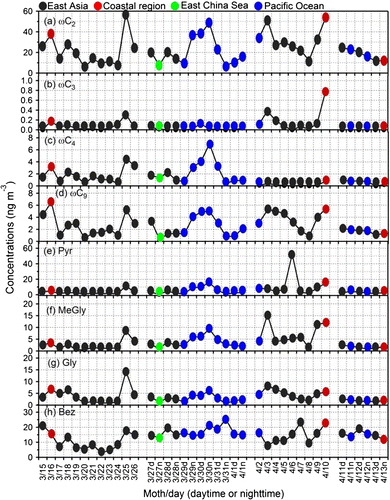
Fig. 4. Temporal variations in the concentration of (a) glyoxylic acid (ωC2), (b) 3-oxopropanoic acid (ωC3), (c) 4-oxobutanoic acid (ωC4), (d) 9-oxononanoic acid (ωC9), (e) pyruvic acid (Pyr), (f) methylglyoxal (MeGly), (g) glyoxal (Gly) and (h) benzoic acid (Bez) in the ambient aerosols from Cape Hedo, Okinawa in spring 2007.
The highest concentration of C2 was observed on April 10 when air masses were delivered from coastal region of China. However, the highest concentrations of C3, C4, pimelic acid (C7) were observed on March 30 (night), when air masses were originated from the Pacific Ocean and travelled over Taiwan before arriving at Okinawa. Highest concentrations of C6 and C9 were observed on March 25 when air masses were delivered from East Asia. Cyclic olifines emitted from vehicles are the source of C6 diacid in the atmosphere (Grosjean et al., Citation1978). The concentration trends of normal chain diacids are similar to each other (Fig. b–g).
Phthalic acid (Ph) is a tracer of anthropogenic emissions (vehicle exhaust) (Kawamura and Ikushima, Citation1993). Terephthalic (tPh) acid is derived from industrial emission and used as major raw material for polyester (polyethylene terephthalate) fibre and plastic bottles (Ding and Li, Citation2003) and is emitted to the atmosphere by plastic burning (Simoneit et al., Citation2005; Kawamura and Pavuluri, Citation2010). Ph, tPh and aliphatic unsaturated diacids (M, mM) show similar trends with higher peaks on 25 April (Fig. h–j). Large amounts of Ph and tPh were found in the Chinese atmosphere (Ho et al., Citation2006; Wang et al., Citation2009). In contrast, fumaric acid (F) showed very high peak on April 11 (day), when air masses were delivered from northeastern China (Fig. k). The concentration trends of oxoacids (glyoxylic, 3-oxopropanoic, 4-oxobutanoic and 9-oxononoic acids) and α-dicarbonyls (MeGly and Gly) are similar each other, suggesting similar sources of these compounds (Fig. ).
On the other hand, pyruvic acid showed a peak on April 6 (Fig. e) when air masses were delivered from East Asia. Pyruvic acid is formed by the oxidation of aromatic hydrocarbons such as toluene and benzene in the atmosphere (Kawamura et al., Citation1996) and its formation is enhanced in the presence of cloud (Ervens et al., Citation2004; Lim et al., Citation2005; Legrand et al., Citation2007). We did not find any clear diurnal trends in 12 h samples for diacids, oxoacids and dicarbonyls (Figs. and ), suggesting that local anthropogenic contribution is not important for these species. Pavuluri et al. (Citation2010) reported a clear day and night-time trend in Chennai, India and Mangshan, China, where local sources and in situ formation of diacids and related compounds are both important. He and Kawamura (Citation2010) also reported similar results.
Figure shows temporal (3 h intervals) variations of selected diacids and oxoacids as well as pyruvic acid, α-dicarbonyls and benzoic acid. ωC2 and α-dicarbonyls show similar temporal variations (Fig. ). Enhanced concentrations of normal diacids (C2–C9, except for C6) and aromatic diacids (Ph, tPh) were observed at 13:30 pm on 14 April and at 16:30 pm on 16 April. For both dates, air mass trajectories demonstrate the influence from East Asia. Lower concentrations of aromatic diacids (Ph and tPh) were observed on 15 April (Fig. h and i) due to the rainfall from 8 am to noon on 15 April. In contrast, higher concentration of fumaric acid (F) was observed on the same date (Fig. g). The peak of F suggests an importance of photo-isomerization of maleic acid (M) under the strong sunlight immediately after the end of rain. The different temporal trend of F from other diacids suggests that source and/or formation mechanism of F is different. Atmospheric NO3− is emitted from anthropogenic sources (Deshmukh et al., Citation2016). Similar trends of diacids and NO3− in 3 h samples suggest that diacids are delivered from East Asia to Okinawa. Individual back trajectories suggest a typical advection time of 2–4 days from the source area.
Fig. 5. Diurnal variations in the concentration of (a) oxalic acid (C2), (b) malonic acid (C3), (c) succinic acid (C4), (d) adipic acid (C6), (e) azelaic acid (C9), (f) maleic acid (M), (g) fumaric acid (F), (h) phthalic acid (Ph), (i) terephthalic acid (tPh), (j) glyoxylic acid (ωC2), (k) 4-oxobutanoic acid (ωC4), (l) 9-oxononoic acid (ωC9), (m) pyruvic acid (Pyr), (n) methylglyoxal (Gly), (o) glyoxal (MeGly) and (p) benzoic acid (Bez) in the ambient aerosols collected at Cape Hedo, Okinawa in spring 2007.
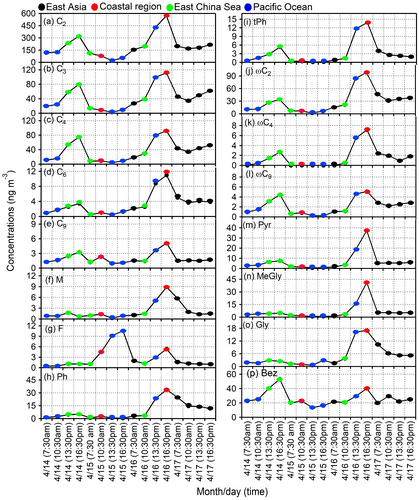
During 15, 16 and 17 March, 5-day back trajectories show that the air masses are originated from East Asia, whereas on 14 March air masses are delivered from the Pacific passing over the southern part of Japan. On 16 April and 14, the 3-h variations of normal diacids (C2–C8), aromatic diacids (Ph and tPh), oxoacids (ωC2), pyruvic acid, MeGly, Gly and benzoic acid all show that the concentrations increased from 10:30 am and peaked at 16:30 pm. The higher concentration of diacids and related compounds at 16:30 is due to increased anthropogenic activities in East Asia via long-range atmospheric transport. However, we did not get any differences in diurnal variation on March 17, although the airmass source regions are East Asia.
3.3. Photochemical processing of diacids and related compounds
Figure shows the average relative abundance (%) of diacid species in total diacids. The average relative abundance of C2 (67%) in this study is much higher than those of malonic (11%) and succinic acid (4%). Very high percentage of C2 indicates a significant photochemical processing of long-chain diacids as well as aqueous phase oxidation of biogenic and anthropogenic volatile organic carbons (VOCs) via Gly, MeGly, Pyr and ωC2 pathway (Ervens et al., Citation2004; Legrand et al., Citation2007), as summarized in Fig. . Relative abundance of individual species is used to evaluate the extent of chemical ageing (He and Kawamura, Citation2010). An increased C2/total diacid ratio (60–70%) has been reported in the marine aerosols from the Pacific during long-range atmospheric transport (Kawamura and Sakaguchi, Citation1999). The relative abundance of C2 from this study (67%) is comparable to those in marine aerosols from the western Pacific (65%) (Wang et al., Citation2006) but less than that from the central equatorial Pacific (>70%) (Kawamura and Sakaguchi, Citation1999).
Fig. 6. Spring time average relative abundance (%) of diacid species in total diacids in aerosols collected at Cape Hedo in 2007.
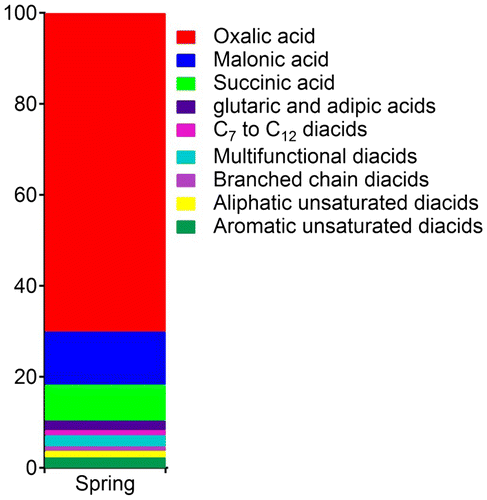
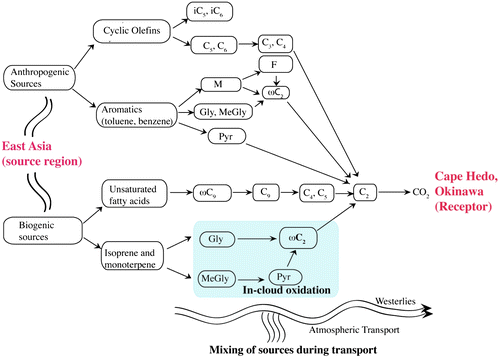
Fig. 7. Schematic diagram of atmospheric aging of selected diacids, oxoacids and α-dicarbonyls at Cape Hedo, Okinawa in the western North Pacific Rim.
For daily and day/night samples, the highest relative abundance of C2 is observed on 24 March (Fig. a) when air masses arrived from East Asia. The average relative abundances of C2 during daytime (70%) and night-time (70%) are similar. There is no sunlight in night-time and the relative humidity is quite high, we suppose that C2 was produced by aqueous phase oxidation of precursors in darkness. Aqueous phase production of C2 in cloud/fog/aerosols is important in the atmosphere (Warneck, Citation2003; Sorooshian et al., Citation2006). Biogenic and anthropogenic VOCs can be oxidized to MeGly and Gly in the gas phase during day and night-time (Kamens et al., Citation1982; Fan and Zhang, Citation2004; Warneck et al., Citation2004). O3 and OH radicals are important oxidants in daytime (Kamens et al., Citation1982; Fan and Zhang, Citation2004), whereas NO3 and H2O2 (in the presence of H2SO4) are main oxidants in night-time (Hermann et al., Citation2000; Claeys et al., Citation2004). NO3 can react with volatile and semi-volatile organic compounds such as hydrated Gly and MeGly in the aqueous phase. Aqueous phase Gly can be oxidized to ωC2, and finally to C2 (Lim et al., Citation2005). Similarly, MeGly is further oxidized to pyruvic acid, hydrated glyoxylic acid and finally to C2 (Legrand et al., Citation2007).
Fig. 8. Temporal variations in the relative abundances of (a) C2/total diacids and C4/total diacids (b) diurnal variations in the relative abundances of C2/total diacids and C4/total diacids in the ambient aerosols collected at Cape Hedo, Okinawa. Colours indicate similar as other figures.
For the 3-h samples, we clearly observed the formation and destruction of short-chain diacids. Figure b shows 3-h variations in relative abundance of oxalic and succinic acids. During the morning hours (centre time 7:30 am) when the relative abundance of C2 is high, the relative abundances of C3 (not shown as a figure) and C4 became low, suggesting that photochemical production of C2 from C3 and C4 is intensified in morning hours. Similarly, when relative abundances of C3 and C4 are significantly higher in the afternoon, relative abundance of C2 is lower, further suggesting the formation of C3 and C4 and/or degradation of C2.
In night-time, oxalic acid is also formed by the degradation of long-chain diacids during long-range atmospheric transport. Figure shows anti-correlations between relative abundances (%) of oxalic acid (C2) in total diacids and concentrations of malonic (C3), succinic (C4), glutaric (C5), adipic (C6) and azelaic (C9) acids. C2 can be produced from long-chain diacids and related compounds (Kawamura and Sakaguchi, Citation1999; Matsunaga et al., Citation1999; Warneck, Citation2003; Carlton et al., Citation2006; Legrand et al., Citation2007, Kundu et al., Citation2010). Previous studies have shown that concentrations of diacids and related compounds inversely correlated with relative abundance of C2 (%) for urban Tokyo (Kawamura and Yasui, Citation2005) and Gosan aerosols (Kundu et al., Citation2010) in summer. In this study, we found that lower concentrations of long-chain diacids are associated with higher relative abundances of C2 (%) (Fig. ). These results suggest that long-chain diacids can be decomposed to C2. Thus, it can be concluded that secondary production and degradation of organic aerosols are more intensified in night-time due to abundant presence of oxidants.
Fig. 9. Scatter plots between relative abundances of oxalic acid (C2/total diacids) and its precursors compounds (a) malonic (C3), (b) succinic (C4), (c) glutaric (C5) (d) adipic (C6), (e) pimelic (C7) and (f) azelaic (C9) acids in night-time during 27 March–1 April and 11–13 April, 2007.
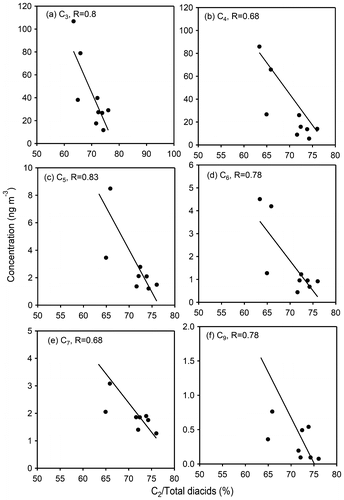
Scatter plots (Fig. S2) between relative abundance of oxalic acid and its precursor compounds such as C3, C4, C5, C6, C7, ωC2, MeGly and Gly further support that oxalic acid is formed by the degradation of long-chain diacids, oxoacids and α-dicarbonyls in early morning hours. We further checked the correlations among OC, C2, C3, C4, C6, Ph, tPh, ωC2 and 9-oxononanoic acid (ωC9). We found very strong correlations among them, suggesting that morning-time aerosols are influenced by long-range atmospheric transport. Large amounts of unsaturated fatty acids are emitted in the Chinese atmosphere from biogenic sources, meat cooking operation and wood burning and degraded to small diacids during long-range transport to Okinawa. We also found that when relative abundance of C4 increases, relative abundance of C2 decreases (Fig. b) in the afternoon, suggesting a favourable degradation of C2 in the presence of Fe2+, during which C2 can be degraded easily via the formation of oxalate-Fe complex to result in formic acid and CO2 (Pavuluri and Kawamura, Citation2012; Zuo and Hoigne Citation1992, Citation1994), although we did not measure Fe in this study.
Oxalic acid–C/OC ratio can be used as a tracer for photochemical aging of organic aerosols (Kunwar and Kawamura, Citation2014b). Figure S3 presents the contribution (%) of selected diacids to organic carbon. Total diacid–C/OC, oxoacid–C/OC and α-dicarbonyl–C/OC ratios ranged from 0.92 to 12% (6.2 ± 2.0%), 0.02 to 1.90% (0.70 ± 0.43%), and 0.03 to 0.48% (0.2 ± 0.09%), respectively. Normal chain diacids (C2 to C5 and C7) show the similar trend. For day and night samples, we did not find clear trends although we got higher ratio in March 27 (day), 28 (night), 29 (night) and 30 (day and night) samples. A very high ratio was observed on 7 April for C2 and ωC2 (Fig. S3a and g). ωC2 is the precursor of C2, which is produced in the atmosphere by the oxidation of aromatic hydrocarbons as well as VOCs such as isoprene (Limbeck et al., Citation2001), terpenes (Fick et al., Citation2004) and xylene (Birdsall and Matthew, Citation2011). Except for a few samples (25, 29 March, 3 and 4 April), the daily and day/night variations of C2 and ωC2 are similar.
Pyruvic acid is formed in the atmosphere by the oxidation of aromatic hydrocarbon (Kawamura et al., Citation1996; Legrand et al., Citation2007). Pyr–C/OC ratios (%) are almost constant except for March 29 (night), 30 (day and night) and April 6 (Fig. S3h). Very high value was observed on 6 April. Gly and MeGly are the oxidation products of anthropogenic VOCs such as naphthalene, alkylnapthalene and toluene as well as biogenic VOCs such as isoprene and terpenes (Kunwar and Kawamura, Citation2014b and reference therein). These semi-volatile precursors of diacids are further oxidized to result in C2. Except for the samples of 17 March and 3 April and 4, Gly shows similar trend with C2 (Fig. S3i).
For 3-h samples, ratios of C2 to C5, ωC2 and benzoic acid increased until 13:30 pm on 14 April and then decreased (Fig. S4). However, C2 peak is not prominent, suggesting that the aerosols from Cape Hedo are more aged. During 15 March, C2–C/OC ratio is sharply decreased at 13:30 pm (Fig. S4a) as well as the ratios of other species. This sharp decrease should be caused by rainfall, suggesting that particles associated with oxalic acid and related water-soluble organic species act as CCN and thus selectively scavenged from the air. There is heavy rainfall in Okinawa before the noontime on that day.
Interestingly, fumaric (F) acid is formed soon after the rain ended on 14 April (Fig. S4e) and solar radiation came back, showing a peak at 16:30 pm. Except for F, all the diacids and related compounds show lower ratio at this time, suggesting the additional source of fumaric acid. tPh, a tracer of plastic burning (Jung et al., Citation2010; Pavuluri et al., Citation2010), shows time specific diurnal variation. Higher values were obtained at 16:30 pm on 14 March and at 13:30 pm and 16:30 pm on 16 April, suggesting the influence from plastic burnings in the afternoon. These results suggest that low molecular weight diacids, oxoacids and α-dicarbonyls are more aged in the Okinawa aerosols during long-range transport from East Asia as illustrated in Fig. .
3.4. Correlation coefficients among selected diacids and related compounds
Correlation analysis was performed separately for day and night samples and for 3-h samples to better understand the chemical processing in the atmosphere. For daytime samples (12 h), C2 is correlated with its precursor compounds such as C3 (r = 0.92), C4 (0.91), and ωC2 (0.94). MSA− that is produced by the atmospheric oxidation of dimethyl sulphide (DMS) and dimethyl sulphoxide (DMSO), shows a strong correlation with C2 (0.75). C2 showed no correlation or weak correlation with NH4+ (0.07), NO3− (0.38), non-sea salt (nss)-SO42− (0.22) and nss-Ca2+ (0.1). No correlation between of C2 and nss-SO42− suggest the different sources or different formation mechanism. In night-time samples, C2 is strongly correlated with its precursors such as C3 (r = 0.91), C4 (0.94), C6 (0.92) and C7 (0.83). Again, C2 is strongly correlated with ωC2 (0.96), Pyr (0.95), Gly (0.86) and MeGly (0.77). Strong correlations of C2 with longer chain diacids suggest that C2 is formed by the oxidation of long-chain diacids. Strong correlations among C2, Pyr, Gly and MeGly further suggest that aqueous phase reaction is also important in night-time during long-range atmospheric transport (Fig. ).
Both nss-SO42− and NO3− are emitted to the atmosphere from the anthropogenic activities including industrial and vehicular emissions and biomass burning (Wang et al., Citation2009). Formation of sulphate from SO2 oxidation is either accomplished in gas phase by OH radicals (homogenous reaction) or through in-cloud/fog processes (heterogeneous pathway) by oxidants. In night-time, we found very high concentration (15 μg m−3) of nss-SO42− on 11 April (Fig. S5). We considered this value as an outlier. After the removal of outlier, we found a good correlation (r = 0.86) between C2 and nss-SO42− in night-time (Fig. S3). A good correlation between these two components suggests the main formation pathways from gaseous precursors in aqueous phase chemistry in night-time. In contrast, no correlation between C2 and nss-SO42− in daytime may demonstrate that local biogenic emission of isoprene, monoterpene and fatty acids are the sources of C2. We further found strong correlation of OC with C2 (r = 0.97), C3 (0.98), C4 (0.98), C6 (0.92), Ph (0.88), ωC2 (0.98), ωC9 (0.96) and Gly (0.94) in night-time.
Interestingly, we could not find such correlations of OC in daytime, except for ωC9 and ωC2. ωC9 is an intermediate compound during the oxidation of biogenic oleic acid (C18:1) to C9 (Kawamura and Gagosian, Citation1987). In daytime, we found very strong correlation of OC with ωC9 and ωC2. These results further suggest that local biogenic emission is important in daytime during 27 March to 1 April and 11–13 April. Tambunan et al. (Citation2006) and Zhu et al. (Citation2016) reported that there are isoprene emitter trees in Okinawa. NO3− shows weak correlation with C2 in daytime (0.38) and strong correlation (0.70) in night-time. The aqueous phase production of oxalic acid in the presence of NO3− may be important in night-time (Harrmann et al., Citation2000). The positive correlations of C2 with NO3− and nss-SO42− in night-time suggest that aqueous phase production of C2 in the presence of H2O2 along with H2SO4. Claeys et al. (Citation2004) reported the aqueous phase chemistry of secondary organic aerosols in the presence of H2O2 and acid.
tPh is a tracer of plastic burning (Simoneit et al., Citation2005; Pavuluri et al., Citation2010). We found a fair correlation between C2 and tPh during night-time (r = 0.61). Plastic burning is significant in East Asia including Mongolia (Jung et al., Citation2010) and influences the study site via long-range atmospheric transport. However, correlation of C2 with tPh is rather weak (0.28) in daytime.
MSA− shows a strong correlation with C2 during daytime (0.75) and night-time (0.88). MSA− can be found in both marine and coastal regions. However, negative correlation of C2 with ss-SO42− suggests an anthropogenic origin. Dimethyl sulphide (DMS) from terrestrial emissions and dimethyl sulphoxide (DMSO) from industrial wastes could be two possible precursors of MSA− (Hui et al., Citation2004).
For the samples collected at 7:30 am, we found a strong correlated of C2 with C3, C4 and C5 (r = 0.80) as well as nss-SO42− (0.93), NH4+ (0.64), NO3− (0.94), MSA− (0.80) and nss-Ca2+ (0.94). Similarly, C2 is strongly correlated with tracers for secondary sources (nss-SO42−, NO3−, MSA− and NH4+) at 10:30 am (0.84, 0.77, 0.62 and 0.64, respectively), 13:30 pm (0.99, 0.91, 0.99 and 0.99, respectively) and 16:30 pm (0.94, 0.87, 0.84 and 0.99, respectively). However, correlation between oxalic acid and benzoic acid is time specific. At 7:30 am, there is negative correlation between C2 and benzoic acid. At 10:30 am, the correlation is very weak (0.09). During afternoon (at 13:30 pm and 16:30 pm), correlations are fair (0.62 and 0.67, respectively), suggesting the common source of C2 and benzoic acid in the afternoon because benzoic acid is emitted from the vehicle exhausts (Ho et al., Citation2010) and local anthropogenic input is less important, hence, we conclude that benzoic acid and its precursors such as toluene were emitted to the Chinese atmosphere from vehicles and long-range transported to Okinawa.
Although daytime (12 h) samples in March and April did not show any correlation between C2 and nss-SO42−, we found a strong correlation in 3-h daytime samples during 14–17 April, suggesting more anthropogenic influence on diacid concentrations. We further checked the concentration of nss-SO42− during this period and concentration of nss-SO42− is very high during the same period except for 15 April. During this period significant amounts of nss-SO42− were emitted to the Chinese atmosphere and long-range transported to the western North Pacific, and thus we detected large amounts of nss-SO42− (up to 30 μg m−3) in Okinawa during this period. Compared to daytime (12 h) samples during late March to mid April, 3 h daytime samples during 14–17 April are more influenced by anthropogenic activities.
3.5. In situ formation of oxalic acid and possible photolysis during long-range atmospheric transport
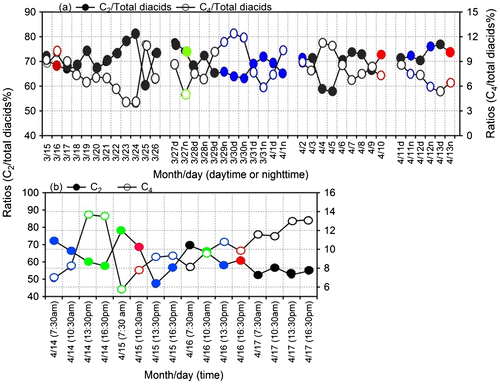
Malonic acid (C3) can be formed in the atmosphere by the photochemical degradation of succinic acid and thus C3/C4 ratio has been used as indicator of enhanced photochemical aging of organic aerosols (Kawamura and Ikushima, Citation1993; Ho et al., Citation2007; Deshmukh et al., Citation2016). Lower ratios (0.25–0.45, av. 0.35) have been reported in vehicular exhaust in comparison to those of atmospheric aerosols (0.6–2.9, av. 1.6) (Ho et al., Citation2007 and reference therein). The C3/C4 ratios ≥ 3 are often reported in aged aerosols (Kundu et al., Citation2010; Kunwar and Kawamura, Citation2014b). In this study, we found similar C3/C4 ratios between 24 h samples (1.54) and day (1.56) and night-time samples (1.57). Interestingly, 3 h samples generally showed lower ratios than daily and day/night samples. The C3/C4 ratios at 7:30 am, 10:30 am, 13:30 pm and 16:30 pm are 1.4, 1.2, 1.0 and 1.1, respectively. The C3/C4 ratios at morning time (7:30) are higher during 14, 15 and 16 March (Fig. d), suggesting that morning-hour aerosols are more aged than other aerosols. The C3/C4 ratios from this study are higher than that (0.61) from summer aerosols in northern Chinese cities (Ho et al., Citation2007).
Fig. 10. Diurnal variations in the concentration ratios of (a) oxalic/malonic (C2/C3), (b) oxalic/succinic (C2/C4), (c) oxalic/glyoxal (C2/Gly), (d) malonic/succinic (C3/C4), (e) oxalic/methylglyoxal (C2/MeGly), (f) oxalic/pyruvic (C2/Pyr) and (g) oxalic/ glyoxylic (C2/ωC2) in the ambient aerosols collected at Cape Hedo, Okinawa.
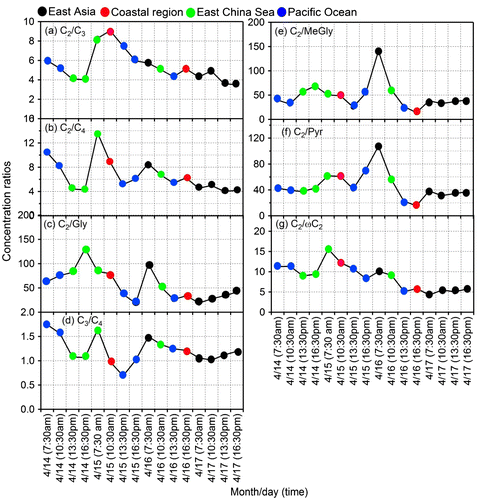
The ratios of C2/C3, C2/C4, C2/MeGly, C2/Pyr and C2/ωC2 are used to decipher photochemical chain reactions in the atmosphere. Interestingly, those ratios are higher at 7:30 am for 3 days (14, 15 and 16 April) (Fig. ), although we did not find the time specific trend on 17 April. The higher ratios at 7:30 am and strong correlations (r = 0.88–0.99) of OC with C2, C3, C4, C6, Ph, ωC2, ωC9, and Gly in both night-time and morning hours, as well as decreased concentrations of the C2 precursors with increased relative abundance of C2/total diacid (%) (Fig. S2), suggest the accumulation of the precursor compounds in the atmosphere in night-time and subsequent photochemical production of oxalic acid immediately after sunrise. Similar trends are also observed for C2/Pyr and C2/ωC2 although peaks are less prominent for C2/Pyr (Fig. f and g). In contrast, C2/Gly ratios showed higher values at 16:30 pm of 14 April and 7:30 am of 16 April. Similar trend was observed for C2/MeGly ratios. These results suggest that photochemical production of LMW diacids is important early in the morning, although morning aerosols can also be influenced by long-range transport.
Relative abundances of C2 for 24 h samples show negative correlations with the relative abundance of C3, C4, C5 and C6 (figure is not presented). This result again suggests a possible formation of oxalic acid from longer chain diacids such as C3 and C4.
3.6. Principal component analysis (PCA) for dicarboxylic acids and related compounds
We performed time series PCA analysis. First, we normalized the data of chemical species using Statistical Package for the Social Sciences (SPSS) software. Table presents a result of PCA for day and night-time and 24 h samples. The number of PCs to extract is determined based on eigenvalue. The component having more than 1 eigenvalue is extracted here. We used a varimax rotation with Kaisher normalization method. We trimmed the missing or <LOD data which is less than 1%, so, there is not a significant change in result.
Table 2. Results of principal component analysis (varimax rotaion with Kaiser Normalization) on concentration of the selected diacids and related compounds and major ions in the aerosols collected in Cape Hedo, Okinawa in spring, 2007.
We found four components in daytime samples, whereas three components were found in night-time and 24 h samples (Table ). High loadings of C2, C3, C4, C6, C7, Ph, ωC2, Pyr, Gly and MeGly were found in Component 1 in daytime, suggesting an influence from anthropogenic emission with photo-oxidation because these diacids are associated with anthropogenic sources, where Gly, MeGly, Pyr and ωC2 are associated with photochemical oxidation in the atmosphere, and Ph is formed by the oxidation of polycyclic aromatic hydrocarbons (PAHs) including naphthalenes (Ho et al., Citation2010 and references therein). Higher loadings of Na+, Cl− and ss-SO42− in component 2 are associated with marine source. In addition, higher loading of nss-Ca2+ in component 2 is beyond our explanation. Higher loadings of NH4+ and nss-SO42− in component 3 suggest an industrial influence and biomass burning of animal excreta. Based on the PCA analyses, we emphasize that anthropogenic and industrial sources and biomass burning of animal dung are important as well as marine inputs over the study site via long-range transport from the Asian Continent in daytime.
In night-time, we detected very strong loadings of C2-C9, Pyr, ωC2, MeGly and MSA− in component 1, which are associated with secondary oxidation. Very high loadings of Na+, Cl− and ss-SO42− in component 2 indicate the marine influence. High loadings of nss-Ca2+ and nss-K+ in component 2 in night-time suggest the mixed sources from marine and continent have influenced Okinawa. nss-SO42− and NH4+ show high loadings in component 3, suggesting that biomass burning and industrial emission are also important in night-time samples. Indeed, higher amount of DMSO is emitted from the Chinese industries to the atmosphere (Hui et al., Citation2004).
For 24 h samples, we found 3 components with higher loadings of C5 and C6, which are associated with anthropogenic activities because both species are tracers of anthropogenic cyclohexene (Kawamura et al., Citation1996). Higher loadings of Ph, tPh, ωC2 and Gly again suggest the photochemical oxidation of aromatic hydrocarbon. Component 1 is associated with secondary formation of small diacids and related compounds from aromatic diacids; components 2 and 3 and are associated with dust influence and marine emission, respectively. The high loading of tPh in component 1 suggests an influence from plastic burning because tPh is a tracer of plastic burning products (Simoneit et al., Citation2005; Jung et al., Citation2010).
4. Summary and conclusions
Daily (24 h), day/night (12 h) and time-resolved (3 h) samples were collected from Cape Hedo, Okinawa to better understand the source and in situ formation of organic acids in the western North Pacific Rim. Oxalic acid (C2) is found as a dominant species followed by malonic (C3) and succinic (C4) acid. We found higher concentration ratios (C2/C4, C2/C3, C3/C4, and C2/ωC2) at 7:30 am, suggesting that organic precursors are accumulated in night-time and then photo-oxidized early in the morning in the presence of sunlight. No clear trends of diacids and related compounds were found in day and night-time, suggesting that there are no local anthropogenic emissions of diacids and related compounds. The strong correlations of C2 with precursor compounds such as C3, C4, C6, Ph, C9, Gly, MeGly and Pyr suggest the formation of low molecular weight diacids via the aqueous phase oxidation of long-chain diacids and other precursors. Further, the average relative abundance of C2 during day and night-time is similar (70%), suggesting that Okinawa aerosols are already aged during long-range atmospheric transport from the Asian Continent. Strong correlation (0.86) between nss-SO42− and oxalic acid during night-time suggests the similar source and/or formation mechanism. However, no correlation between nss-SO42− and oxalic acid and strong correlation of OC with ωC2 (0.81) and ωC9 (0.73) further suggest the local biogenic emission during daytime. For 3-h samples, oxalic acid shows good correlations with secondary atmospheric tracers (NO3−, SO42−, NH4+, MSA−), again supporting the secondary photochemical production of C2 during long-range atmospheric transport. Compared with daytime (12 h) samples on 27 March–1 April and 11–13 April, we found that 3-h daytime samples on 14–17 April are more influenced by anthropogenic activities in China via long-range transport.
Disclosure statement
No potential conflict of interest was reported by the authors.
Funding
This study was in part supported by the Japan Society for the Promotion of Science [grant-in-aid number 1920405], [grant-in-aid number 24221001]; by the Environment Research and Technology Development Fund (B-0903) from the Ministry of the Environment, Japan.
Supplemental data
Supplemental data for this article can be accessed here https://doi.org/10.1080/16000889.2017.1369341.
Supplementary Material
Download MS Word (7 MB)Acknowledgements
We thank S. G. Aggarwal, Y. Kitamori, K. Okuzawa, M. Mochida and C. Nishida for the help in aerosol sampling at Cape Hedo, Okinawa, Japan and A. Takami of the National Institute of Environmental Studies, Japan for the courtesy to use the CHAARM site at Cape Hedo. We also thank the NOAA Air Resources Laboratory (ARL) for the provision of the HYSPLIT transport model and READY website (https://www.arl.noaa.gov/ready.php) used in this publication.
References
- Birdsall, A. W. and Matthew, E. J. 2011. Comprehensive NO-dependent study of the products of the oxidation of atmospherically relevant aromatic compounds. J. Phys. Chem. 115, 5397–5407.10.1021/jp2010327
- Carlton, A. G., Turpin, B. J., Lim, H. J., Altieri, K. E. and Seitzinger, S. 2006. Link between isoprene and secondary organic aerosol (SOA): Pyruvic acid oxidation yields low volatility organic acids in clouds. Geophys. Res. Lett. 33, DOI:10.1029/2005GL025374.
- Claeys, M., Wang, W., Ion, A. C., Kourtchev, I., Gelencsér, A. and co-authors. 2004. Formation of secondary organic aerosols from isoprene and its gas-phase oxidation products through reaction with hydrogen peroxide. Atmos. Environ. 38, 4093–4098.10.1016/j.atmosenv.2004.06.001
- Deshmukh, D. K., Kawamura, K., Lazaar, M., Kunwar, B. and Boreddy, S. K. R. 2016. Dicarboxylic acids, oxoacids, benzoic acid, α-dicarbonyls, WSOC, OC and ions in spring aerosols from Okinwa Island in the western North Pacific Rim: Size distributions and formation processes. Atmos. Chem. Phys. 16, 5263–5282.10.5194/acp-16-5263-2016
- Ding, M. and Li, X. 2003. About the construction of PTA. Chem. Technol. Econ. 21, 13–15.
- Draxler, R. R. and Hess, G. D. 1998. An overview of the HYSPLIT_4 modelling system for trajectories, dispersion and deposition. Aust. Meteorol. Mag. 47, 295–308.
- Duan, J., Tan, J., Yang, L., Wu, S. and Hao, J. 2008. Concentration, sources and ozone formation potential of volatile organic compounds (VOCs) during ozone episode in Beijing. Atmos. Res. 88, 25–35. DOI:10.1016/j.atmosres.2007.09.004.
- Ervens, B., Feingold, G., Frost, G. J. and Kreidenweis, S. M. 2004. A modelling study of aqueous production of dicarboxylic acids: 1. Chemical pathways and speciated organic mass production. J. Geophys. Res. 109, 2247. DOI:10.1029/2003JD004387.
- Fan, J. W. and Zhang, R. Y. 2004. Atmospheric oxidation mechanism of isoprene. Environ. Chem. 1, 140–149.10.1071/EN04045
- Fick, J., Nilsson, C. and Andersson, B. 2004. Formation of oxidation products in a ventilation system. Atmos. Environ. 38, 5895–5899.10.1016/j.atmosenv.2004.08.020
- Fu, P., Kawamura, K., Usukura, K. and Miura, K. 2013. Dicarboxylic acids, ketocarboxylic acids and glyoxal in the marine aerosols collected during a round-the-world cruise. Mar. Chem. 148, 22–32.10.1016/j.marchem.2012.11.002
- Grosjean, D., Cauwenberghe, K. V., Schmid, J. P., Kelley, P. E. and Pitts, J. N. 1978. Identification of C3–C10 aliphatic dicarboxylic acids in airborne particulate matter. Environ. Sci. Technol. 12, 313–317. DOI:10.1021/es60139a005.
- Hatakeyama, S., Murano, K., Sakamaki, F., Mukai, H., Bandow, H. and co-authors. 2001. Transport of atmospheric pollutants from East Asia. Water Air Soil Pollut. 130, 373–378.10.1023/A:1013877000169
- He, N. and Kawamura, K. 2010. Distributions and diurnal changes of low molecular weight organic acids and α-dicarbonyls in suburban aerosols collected at Mangshan, North China. Geochem. J. 44, e17–e22.10.2343/geochemj.1.0093
- Herrmann, H., Ervens, B., Jacobi, H.-W., Wolke, R., Nowacki, P. and co-authors. 2000. CAPRAM 2.3: A chemical aqueous phase radical mechanism for tropospheric chemistry. J. Atmos. Chem. 36, 231–284.10.1023/A:1006318622743
- Highwood, E. J. and Kinnersley, R. P. 2006. When smoke gets in our eyes: The multiple impacts of atmospheric black carbon on climate, air quality and health. Environ. Int 32, 560–566.10.1016/j.envint.2005.12.003
- Ho, K. F., Cao, J. J., Lee, K. S. C., Kawamura, K., Zhang, J. and co-authors. 2007. Dicarboxylic acids, ketocarboxylic acids, and dicarbonyls in the urban atmosphere of China. J. Geophys. Res. 112, D22S27. DOI:10.1029/2006JD008011.
- Ho, K. F., Lee, S. C., Cao, J. J., Kawamura, K., Watanabe, T., Cheng, Y. and Chow, J. C. 2006. Dicarboxylic acids, ketocarboxylic acids and dicarbonyls in the urban roadside area of Hong Kong. Atmos. Environ. 40, 3030–3040.
- Ho, K. F., Lee, S. C., Ho, S. S. H., Kawamura, K., Tachibana, E. and co-authors. 2010. Dicarboxylic acids, ketocarboxylic acids, α-dicarbonyls, fatty acids, and benzoic acid in urban aerosols collected during the 2006 Campaign of Air Quality Research in Beijing (CAREBeijing-2006). J. Geophys. Res. 115, 1451. DOI:10.1029/2009JD013304.
- Hopke, P. K. 1985. Receptor Modelling in Environmental Chemistry Wiley, New York.
- Hui, Y., Wang, Y. and Zhuang, G. 2004. MSA in Beijing aerosol. Chin. Sci. Bull. 49, 1020–1025.
- Jung, J., Tsatsral, B., Kim, Y. J. and Kawamura, K. 2010. Organic and inorganic aerosol compositions in Ulaanbaatar, Mongolia, during the cold winter of 2007 to 2008: Dicarboxylic acids, ketocarboxylic acids, and α-dicarbonyls. J. Geophys. Res. 115, D03205. DOI:10.1029/2010JD014339.
- Kamens, R. M., Gery, M. W., Jeffries, H. E., Jackson, M. and Cole, E. I. 1982. Ozone-isoprene reactions; Product formation and aerosol potential. Int. J. Chem. Kinet. 14, 955–975.10.1002/(ISSN)1097-4601
- Kawamura, K. and Bikkina, S. 2016. A review of dicarboxylic acids and related compounds in atmospheric aerosols: Molecular distributions, sources and transformation. Atmos. Res. 170, 140–160.10.1016/j.atmosres.2015.11.018
- Kawamura, K. and Gagosian, R. B. 1987. Implications of ω-oxocarboxylic acids in the remote marine atmosphere for photo-oxidation of unsaturated fatty acids. Nature 325, 330–332.10.1038/325330a0
- Kawamura, K. and Ikushima, K. 1993. Seasonal changes in the distribution of dicarboxylic acids in the urban atmosphere. Environ. Sci. Technol. 27, 2227–2235.10.1021/es00047a033
- Kawamura, K., Kasukabe, H. and Barrie, L. A. 1996. Source and reaction pathways of dicarboxylic acids, ketoacids and dicarbonyls in arctic aerosols: One year of observations. Atmos. Environ. 30, 1709–1722.10.1016/1352-2310(95)00395-9
- Kawamura, K., Kasukabe, H. and Barrie, L. A. 2010. Secondary formation of water-soluble organic acids and α-dicarbonyls and their contribution to total carbon and water-soluble organic carbon: Photochemical ageing of organic aerosols in the Arctic spring. J. Geophys. Res. 115, 2542. DOI:10.1029/2010JD014299.
- Kawamura, K., Ng, L.-L. and Kaplan, I. R. 1985. Determination of organic acids (C1–C10) in the atmosphere, motor exhausts, and engine oils. Environ. Sci. Technol 19, 1082–1086.10.1021/es00141a010
- Kawamura, K. and Pavuluri, C. M. 2010. New directions: Need for better understanding of plastic waste burning as inferred from high abundance of terephthalic acid in South Asian aerosols. Atoms. Environ. 44, 5320–5321.10.1016/j.atmosenv.2010.09.016
- Kawamura, K. and Sakaguchi, F. 1999. Molecular distributions of water soluble dicarboxylic acids in marine aerosols over the Pacific Ocean including tropic. J. Geophys. Res. 104, 3501–3509.10.1029/1998JD100041
- Kawamura, K., Steinberg, S. and Kaplan, I. R. 1985. Capillary GC determination of short-chain dicarboxylic acids in rain, fog, and mist. Int. J. Environ. Anal. Chem. 19, 175–188.10.1080/03067318508077028
- Kawamura, K., Tachibana, E., Okuzawa, K., Aggarwal, S. G., Kanaya, Y. and co-authors. 2013. High abundances of water-soluble dicarboxylic acids, ketocarboxylic acids and α-dicarbonyls in the mountaintop aerosols over the North China Plain during wheat burning season. Atmos. Chem. Phys. 13, 8285–8302.10.5194/acp-13-8285-2013
- Kawamura, K. and Yasui, O. 2005. Diurnal changes in the distribution of dicarboxylic acids, ketocarboxylic acids and dicarbonyls in the urban Tokyo atmosphere. Atmos. Environ. 39, 1945–1960.10.1016/j.atmosenv.2004.12.014
- Kundu, S., Kawamura, K. and Lee, M. 2010. Seasonal variations of diacids, ketoacids and α-dicarbonyls in marine aerosols at Gosan, Jeju Island: Implications for their formation and degradation during long-range transport. J. Geophys. Res. 115, 1046. DOI:10.1029/2010JD013973.
- Kunwar, B. and Kawamura, K. 2014a. One-year observations of carbonaceous and nitrogenous components and major ions in the aerosols from subtropical Okinawa Island, an outflow region of Asian dusts. Atmos. Chem. Phys. 14, 1819–1836.10.5194/acp-14-1819-2014
- Kunwar, B. and Kawamura, K. 2014b. Seasonal distributions and sources of low molecular weight dicarboxylic acids, ω-oxocarboxylic acids, pyruvic acid, α-dicarbonyls and fatty acids in ambient aerosols from subtropical Okinawa in the western Pacific Rim. Environ. Chem. 11, 673–689.10.1071/EN14097
- Kunwar, B., Kawamura, K. and Zhu, C. 2016. Stable carbon and nitrogen isotopic compositions of ambient aerosols collected from Okinawa Island in the western North Pacific Rim, an outflow region of Asian dusts and pollutants. Atmos. Environ. 131, 243–253.10.1016/j.atmosenv.2016.01.035
- Kunwar, B., Torii, K., Zhu, C., Fu, P. and Kawamura, K. 2016. Springtime variations of organic and inorganic constituents in submicron aerosols (PM1.0) from Cape Hedo, Okinawa. Atmos. Environ. 130, 84–94.10.1016/j.atmosenv.2015.09.002
- Legrand, M., Preunkert, S., Oliveira, T., Pio, C. A., Hammer, S. and co-authors. 2007. Origin of C2–C5 dicarboxylic acids in the European atmosphere inferred from year-around aerosol study conducted at a west-east transect. J. Geophys. Res. 112, D23S07. DOI:10.1029/2006JD008019.
- Lim, H.-J., Carlton, A. G. and Turpin, B. J. 2005. Isoprene forms secondary organic aerosol through cloud processing: Model simulations. Environ. Sci. Technol. 39, 4441–4446.10.1021/es048039h
- Limbeck, A., Puxbaum, H., Otter, L. and Scholes, M. C. 2001. Semivolatile behavior of dicarboxylic acids and other polar organic species at a rural background site (Nylsvley, RSA). Atmos. Environ. 35, 1853–1862.10.1016/S1352-2310(00)00497-0
- Matsunaga, S., Kawamura, K., Nakatsuka, T. and Ohkouchi, N. 1999. Preliminary study on laboratory photochemical formation of low weight dicarboxylic acids from unsaturated fatty acid (oleic acid). Res. Org. Geochem. 14, 19–25.
- Mochida, M., Kawabata, A., Kawamura, K., Hatsushika, H. and Yamazaki, K. 2003. Seasonal variation and origins of dicarboxylic acids in the marine atmosphere over the western North Pacific. J. Geophys. Res. 108, 4193. DOI:10.1029/2002JD002355.
- Novakov, T. and Penner, J. E. 1993. Large contribution of organic aerosols to cloud-condensation-nuclei concentrations. Nature 365, 823–826.10.1038/365823a0
- Ohara, T., Akimoto, H., Kurokawa, J., Horii, N., Yamaji, K. and co-authors. 2007. An Asian emission inventory of anthropogenic emission sources for the period 1980–2020. Atmos. Chem. Phys. 7, 4419–4444.10.5194/acp-7-4419-2007
- Pavuluri, C. M. and Kawamura, K. 2012. Evidence for 13-carbon enrichment in oxalic acid via iron catalyzed photolysis in aqueous phase. Geophys. Res. Lett. 39, L03802. DOI:10.1029/2011GL050398.
- Pavuluri, C. M., Kawamura, K. and Swaminathan, T. 2010. Water-soluble organic carbon, dicarboxylic acids, ketoacids, and α-dicarbonyls in the tropical Indian aerosols. J. Geophys. Res. 115, D11302. DOI:10.1029/2009JD012661.
- Pokhrel, A., Kawamura, K., Ono, K., Seki, O., Fu, P. and co-authors. 2016. Ice core records of monoterpene- and isoprene-SOA tracers from Aurora Peak in Alaska since 1660s: Implication for climate change variability in the North Pacific Rim. Atmos. Environ. 130, 105–112.10.1016/j.atmosenv.2015.09.063
- Pokhrel, A., Kawamura, K., Seki, O., Matoba, S. and Shiraiwa, T. 2015. Ice core profiles of saturated fatty acids (C12:0–C30:0) and oleic acid (C18:1) from southern Alaska since 1734 AD: A link to climate change in the Northern Hemisphere. Atmos. Environ. 100, 202–209.10.1016/j.atmosenv.2014.11.007
- Sato, K., Li, H., Tanaka, Y., Ogawa, S., Iwasaki, Y. and co-authors. 2009. Long-range transport of particulate polycyclic aromatic hydrocarbons at Cape Hedo remote island site in the East China Sea between 2005 and 2008. J. Atmos. Chem. 61, 243–257.
- Saxena, P., Hildemann, L., McMurry, P. and Seinfeld, J. 1995. Organics alter hygroscopic behavior of atmospheric particles. J. Geophys. Res. 100, 18755–18770.10.1029/95JD01835
- Sempéré, R. and Kawamura, K. 2003. Trans-hemispheric contribution of C2–C10 α, ω-dicarboxylic acids, and related polar compounds to water-soluble organic carbon in the western Pacific aerosols in relation to photochemical oxidation reactions. Global Biogeochem. Cycles 17, 1069. DOI:10.1029/2002GB001980.
- Simoneit, B. R. T., Medeiros, P. M. and Didyk, B. M. 2005. Combustion products of plastics for refuse burning in the atmosphere. Environ. Sci. Technol. 39, 6961–6970.10.1021/es050767x
- Sorooshian, A., Varutbangkul, V., Brechtel, F. J., Ervens, B., Feingold, G. and co-authors. 2006. Oxalic acid in clear and cloudy atmospheres: analysis of data from International Consortium for Atmospheric Research on Transport and Transformation 2004. J. Geophys. Res. Atmos. 111, D22. DOI:10.1029/2005jd006880.
- Suh, I., Zhang, R., Molina, L. T. and Molina, M. J. 2003. Oxidation mechanism of aromatic peroxy and bicyclic radicals from OH−Toluene reactions. J. Am. Chem. Soc. 125, 12655–12665. DOI:10.1021/ja0350280.
- Takami, A., Miyoshi, T., Shimono, A., Kaneyasu, N., Kato, S. and co-authors. 2007. Transport of anthropogenic aerosols from Asia and subsequent chemical transformation. J. Geophys. Res. 112, D22S31. DOI:10.1029/2006jd008120.
- Tambunan, P., Baba, S., Kuniyoshi, A., Iwasaki, H., Nakamura, T. and co-authors. 2006. Isoprene emission from tropical trees in Okinawa Island, Japan. Chemosphere 65, 2138–2144.10.1016/j.chemosphere.2006.06.013
- Ueda, S., Osada, K. and Takami, A. 2011. Morphological features of soot-containing particles internally mixed with water-soluble materials in continental outflow observed at Cape Hedo, Okinawa. Japan. J. Geophys. Res. 116, 6469. DOI:10.1029/2010JD015565.
- Wang, H., Kawamura, K. and Shooter, D. 2005. Carbonaceous and ionic components in wintertime atmospheric aerosols from two New Zealand cities: Implication for solid fuel combustion. Atmos. Environ. 39, 5865–5875.10.1016/j.atmosenv.2005.06.031
- Wang, H., Kawamura, K. and Yamazaki, K. 2006. Water soluble dicarboxylic acids, ketoacids and dicarbonyls in the atmospheric aerosols over the Southern Ocean and Western North Pacific. J. Atmos. Chem. 53, 43–61.10.1007/s10874-006-1479-4
- Wang, G. H., Kawamura, K., Umemoto, N., Xie, M. J., Hu, S. Y. and co-authors. 2009. Water-soluble organic compounds in PM2.5 and size-segregated aerosols over Mount Tai in North China Plain. J. Geophys. Res. 114, D19208. DOI:10.1029/2008jd011390.
- Wang, G., Xie, M., Hu, S., Gao, S., Tachibana, E. and Kawamura, K. 2010. Dicarboxylic acids, metals and isotopic compositions of C and N in atmospheric aerosols from inland China: implications for dust and coal burning emission and secondary aerosol formation. Atmos. Chem. Phys. 10, 6087–6096.
- Warneck, P. 2003. In-cloud chemistry opens pathway to the formation of oxalic acid in the marine atmosphere. Atmos. Environ. 37, 2423–2427.10.1016/S1352-2310(03)00136-5
- Warneke, C., de Gouw, J. A., Goldan, P. D., Kuster, W. C., Williams, E. J. and co-authors. 2004. Comparison of daytime and nighttime oxidation of biogenic and anthropogenic VOCs along the New England coast in summer during New England Air Quality Study 2002. J. Geophys. Res. 109, 2169–2897. DOI:10.1029/2003jd004424.
- Yamamoto, S. and Kawamura, K. 2011. Stable hydrogen isotopic compositions of fossil fuel-derived n-alkanes in the atmospheric aerosols from Okinawa. Jpn. Res. Org. Geochem. 44, 81–89.
- Zhu, C., Kawamura, K. and Fu, P. 2016. Seasonal variations of biogenic secondary organic aerosol tracers in Cape Hedo, Okinawa. Atmos. Environ. 130, 113–119.10.1016/j.atmosenv.2015.08.069
- Zuo, Y. G. and Hoigne, J. 1992. Formation of hydrogen-peroxide and depletion of oxalic-acid in atmospheric water by photolysis of iron(III) oxalato complexes. Environ. Sci. Technol. 26, 1014–1022.10.1021/es00029a022
- Zuo, Y. G. and Hoigne, J. 1994. Photochemical decomposition of oxalic, glyoxalic and pyruvic-acid catalyzed by iron in the atmospheric waters. Atmos. Environ. 28, 1231–1239.10.1016/1352-2310(94)90270-4