
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
This study presents and evaluates the most comprehensive set to date of chemical, physical and optical properties of aerosols in the outflow from South Asia covering a full winter (Nov. 2014 – March 2015), here intercepted at the Indian Ocean receptor site of the Maldives Climate Observatory in Hanimaadhoo (MCOH). Cluster analysis of air-mass back trajectories for MCOH, combined with AOD and meteorological data, demonstrate that the wintertime northern Indian Ocean is strongly influenced by aerosols transported from source regions with three major wind regimes, originating from the Indo-Gangetic Plain (IGP), the Bay of Bengal (BoB) and the Arabian Sea (AS). As much as 97 ± 3% of elemental carbon (EC) in the PM10 was also found in the fine mode (PM2.5). Other mainly anthropogenic constituents such as organic carbon (OC), non-sea-salt (nss) -K+, nss-SO42− and NH4+ were also predominantly in the fine mode (70–95%), particularly in the air masses from IGP. The combination at this large-footprint receptor observatory of consistently low OC/EC ratio (2.0 ± 0.5), strong linear relationships between EC and OC as well as between nss-K+ and both OC and EC, suggest a predominance of primary sources, with a large biomass burning contribution. The particle number-size distributions for the air masses from IGP and BoB exhibited clear bimodal shapes within the fine fraction with distinct accumulation (0.1 μm < d < 1 μm) and Aitken (0.025 μm < d < 0.10 μm) modes. This study also supports that IGP is a key source region for the wider South Asia and nearby oceans, as defined by the criteria that anthropogenic AODs exceed 0.3 and absorption AOD > 0.03. Taken together, the aerosol pollution over the northern Indian Ocean in the dry season is dominated by a well-mixed long-range transported regime of the fine-mode aerosols largely from primary combustion origin.
1. Introduction
Aerosol loadings in the South Asian atmosphere are much higher than the global average, largely due to anthropogenic activities. Black Carbon aerosols (BC, also known as soot) originate from incomplete combustion of biomass and fossil fuels. BC is an important component of Atmospheric Brown Clouds (ABC), which are visible from space, spreading out from India, and other key source regions. Given the relatively short atmospheric lifetime of BC (~ 1–2 weeks), the climate effects of BC are more regional compared to those of long-lived gases such as CO2 (atmospheric lifetime 100 – 1000s of years). It is therefore important to investigate BC in the regions where the climate effects are the highest. In addition to having a strong regional impact on the South Asian climate (e.g., Satheesh and Ramanathan, Citation2000; Lawrence and Lelieveld, Citation2010; Bond et al., Citation2013; Boucher, et al., Citation2013), BC particles also have adverse effects on human health (Janssen et al., Citation2012; WHO, Citation2016), agriculture and precipitation patterns (e.g., Ramanathan et al., Citation2001), and the melting of snow and ice in the Himalaya-Tibetan plateau (e.g., Menon et al., Citation2010; Kaspari et al., Citation2013; Li et al., Citation2016).
While the climate implications of BC and other anthropogenic aerosols in South Asia are considered to be of global importance, there are still many large uncertainties. BC is co-emitted with a variety of other aerosols and aerosol precursor gases. Thus, the knowledge about particle number and size distribution, chemical composition and mixing ratios of different species is essential for developing the basic understanding of atmospheric processes that affect the composition and hence the effects of South Asian aerosols.
There is only one earlier report with a comprehensive coverage of core aerosol parameters through a winter campaign from the northern Indian Ocean (Ramanathan et al., Citation2007a). In addition to the need to compare with that pioneering 2004–2005 study, also held at MCOH, and to add a second set of observations, the current study is performed ten years later in this region with presumably rapidly changing emission scenarios. The current study also adds several additional dimensions compared to the 2004–2005 investigations. For instance, our new study provides information on the relative importance of fine (PM2.5) versus coarse (PM10–2.5) partitioning of organic carbon (OC), elemental carbon (EC), mass and inorganic aerosol constituents,. Additionally, absorption aerosol optical depth (AAOD) and carbon monoxide (CO) serve as further markers of incomplete combustion. As espoused below, this study thus adds an important decennial revisit of this important aerosol regime expands with several new parameters and establishes both some similarities and some differences between the winter-long campaigns performed ten years apart.
The main objectives of the present study were to provide a comprehensive perspective on the physical, chemical and optical properties of aerosols over the northern Indian Ocean during the six-month-long dry season with highest regional aerosol optical depth (AOD), with specific focus on the homogeneity/regional mixing state, sources and distributional mode of aerosols – fundamental aspects needed to move towards an improved understanding of aerosol dynamics and effects in this high loading – less studied system.
2. Measurements and methods
2.1 The Maldives climate observatory Hanimaadhoo
The location of the MCOH was selected, in the wake of the Indian Ocean Experiment (INDOEX) programme, to intercept the outflow from continental S. Asia with minimal local influence. MCOH is located in the northernmost atoll of the Maldives, on the northernmost of its major inhabited islands, and at the northern point of the 6000 x 700 m sized island of Hanimaadhoo (6.77 °N, 73.18 °E) (Fig. ).
Fig. 1. This map depicts a MODIS satellite image, illustrating the typical high loadings of aerosols (aerosol optical depth-AOD at 550 nm, November 2014 to April 2015) over South Asia. Clustered 6-hourly, 7-day air mass back trajectories (AMBTs) means for Hanimaadhoo (total n = 724). The cluster means lines coloured in map indicate three different source regions: Indo Gangetic Plain (red), Bay of Bengal (yellow) and Arabian Sea (blue).
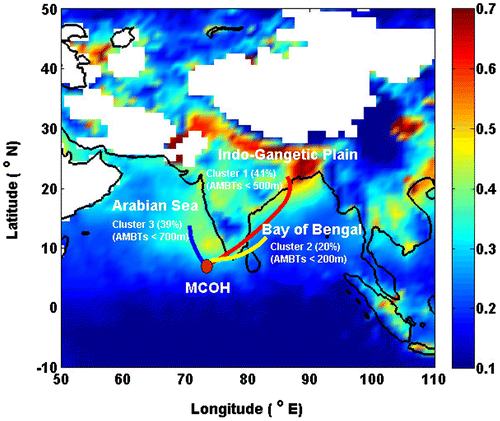
The winter monsoon is typically active from November to April during which NE and NW winds generally bring pollutants from the S. Asian continent to the Indian Ocean (Fig. ). The MCOH laboratory is located 1.5 m above sea level and is surrounded by ocean within 100 m on the W, N and E sides. MCOH has a 15 m atmospheric tower that rises above the local vegetation to allow unobstructed aerosol sampling and solar radiation measurements at the top platform. Various additional on-line measurements and samples are taken in the temperature- and humidity-controlled instrument room through a 15 m long PM10 sampling inlet. More details about the MCOH sampling site and operation of the station are described elsewhere (e.g., Corrigan et al., Citation2006; Budhavant et al., Citation2015a).
2.2 Instrumentation
This study comprises aerosol data from MCOH for six months (November 2014 – April 2015). The instrumentation at the measurement site that is relevant to this work is described below.
We measured the particle number size distributions in a size range of 0.007 – 0.80 μm with a 10-min time resolution using a Differential Mobility Particle Sizer (DMPS). The size-selected particles were detected with a condensation particle counter (CPC, TSI Inc., model 3010) after passage through a 28.5 cm long differential mobility analyzer (DMA). Size selected particles were measured in 35 increments. A neutralizer was used to obtain a steady-state charge distribution of the particles. In addition, total particle number concentrations were measured with another CPC (TSI Inc., model 3772). The integrated total number concentration from the DMPS agreed well (± 5%) with the concentration measured by the standalone CPC.
Concentrations of carbon monoxide (CO) was measured continuously with an APMA-370 Analyzer (Horiba Instruments). The device employs an independent, internal dry-method to achieve a high level of sensitivity and accuracy (± 1% of full scale). The sample is pumped through an oxidizing catalyst that converts the CO to CO2 followed by non-dispersive infrared detection. This sampler has a detection limit of 0.02 ppm (3 times the standard deviation) and the calibration is auto-adjusted regularly with an external CO reference gas.
We also analysed the columnar aerosol optical depth (AOD), single scattering albedo (SSA, ratio of scattering efficiency to total extinction efficiency) and absorption aerosol optical depth (AAOD = [1- SSA] x AOD) with data obtained from the sun photometer, which is part of the AERONET (AErosol RObotic NETwork) program. Here, we used the pre- and post-deployment calibration to attain the quality assured level 2.0 data (almucantar retrieval data); calibrated sky radiance measurements typically have an uncertainty less than 5% (Holben et al., Citation1998, Citation2001). The AOD is representative of columnar aerosol loading, and was here assessed both temporally through a sun photometer placed at the MCOH throughout the campaign, as well as average conditions for Nov 2014–April 2015 over the whole region (Fig. ).
Additionally, a total number of 52 aerosol filter samples (26 of PM2.5 and 26 of PM10) were collected in parallel using two identical high-volume samplers (Digitel AG, Switzerland; model DH-77) that were set up on the top platform of the 15 m MCOH tower, and operating at 500 LPM. All samples were collected on pre-weighed and pre-combusted (5 h at 450 °C) 150 mm diameter quartz filters. Approximately one-week-long collections were performed to enable several elemental, molecular and isotopic analyses in this and forthcoming studies. All the filter handling and analysis of the carbonaceous aerosols followed the quality-controlled analytical protocols described earlier (e.g., Gustafsson et al., Citation2009; Sheesley et al., Citation2012; Bosch et al., Citation2014; Budhavant et al., Citation2015a).
2.3 Chemical analyses
We determined the mass concentrations of elemental carbon (EC) and organic carbon (OC) on the thermal-optical transmission (TOT) based Carbon Analyser from the Sunset Laboratory (Tigard, OR, USA) using the National Institute for Occupational Safety and Health (NIOSH) 5040 method (Birch and Cary, Citation1996). All reported OC results were blank subtracted using an average field filter blank (n = 6). The OC blank was 3% of the average filter loading. No EC was detected in the field blanks. Before the analysis of each batch of samples, the instruments were calibrated in the range 10–50 μgC with sucrose solutions for assessing the accuracy of TC (i.e., OC + EC) and is found to be within 5%. Further, we also analyse a sample which is traceable to the NIST RM-8785 reference material, to ensure that the split between OC and EC is properly estimated. Triplicate analysis was done roughly every sixth filter. The average relative standard deviation of triplicate analysis was 4% for EC and 5% for OC.
An aliquot of each aerosol sample was also extracted with ultrapure deionized water (Milli-Q; Specific resistivity > 18.2 MΩ-cm) and analysed for water-soluble inorganic ionic species (cations: NH4+, Na+, K+, Mg2+, and Ca2+; anions: Cl−, NO3− and SO42−) using a DIONEX Ion Chromatograph (IC; ICS-2000 model). Analytical quality checks were performed with external standards/reference material both with the Sunset OCEC (NIST-SRM urban dust, and gravimetrically prepared solutions of sucrose and potassium hydrogen phthalate) and with the IC (high-purity inorganic salt solutions containing the measured water-soluble inorganic constituents). Random error for the IC analyses, measured with a synthetic sample in each batch, given as one standard deviation, was 10% for cations and 5% for anions. To investigate the effect of marine influences on the aerosol composition, sea salt ratios were calculated using Na+ as the reference element (Keene et al., Citation1986).
2.4 Back-trajectory analysis
Air mass back trajectories were used to identify the pollutant source regions and subsequent transport pathways to the measurement site. The NOAA HYSPLIT (Hybrid Single-Particle Lagrangian Integrated Trajectory (Stein et al., Citation2016) model was used to calculate seven-day air mass back trajectories (at 06:00 h, local time) at an arrival height of 100 m over MCOH (Fig. S1). The trajectory calculations were based on data from the Global Data Assimilation System (GDAS, 1 degree) of the US National Weather Service’s National Centre for Environmental Prediction (NCEP), (Kanamitsu, Citation1989). Transport on local scales (<10 km) surrounding a site is better represented by local surface wind observations, which were obtained by a Vaisala weather station on Hanimaadhoo (Fig. S2). The back trajectories and local measured wind direction showed good agreement with predominantly northeasterly winds. We have also computed the average concentration and diagnostic mass ratios of specific chemical constituents, weighted according to their mean back trajectory paths over MCOH using the concentration-weighted-trajectory analysis (CWT), following a mathematical expression similar to that described earlier (Sheesley et al., Citation2012; Kirillova et al., Citation2013; Bikkina et al., Citation2016).
where wji is the fraction wind originating from source cluster ‘j’ for sample number ‘i’, Ci denotes the concentration of chemical constituents in PM, and CSRj is the mean concentration for cluster ‘j’.
The local meteorological conditions (wind speed and direction, temperature, relative humidity and rainfall), recorded by the Maldives Meteorological Services (MMS) at the Hanimaadhoo airport (1.5 km to the south of the MCOH station), associated with these periods are shown in Figs. S2 and S3. The daily mean relative humidity was 80% and varied in the range 70–93%. The daily mean temperature was 28 °C and varied in the range 25–33 °C.
3. Results and discussion
3.1 Atmospheric transport
The cluster analysis of the HYSPLIT air mass back trajectory files yielded that the air parcels sampled over MCOH during November 2014 – April 2015 could be grouped into three classes. These originated from the Indo-Gangetic Plain (hereafter referred as IGP-origin, red colour), the Bay of Bengal (BoB-origin, yellow) and the Arabian Sea (AS-origin, blue) (Fig. and Fig. S1). About 40% each of the received air parcels at MCOH were classified as IGP-origin and AS-origin with the remaining 20% occurrences were BoB-origin (Fig. ). The spatial distribution of the period-average AOD also demonstrates that these air masses are instrumental in spreading out the aerosol pollution from these key continental source regions to the northern Indian Ocean (Fig. ).
The Indo-Gangetic Plain (IGP) is a 255-million-hectar fertile plain encompassing most of the densely populated and industrialized northern and northeastern India, the eastern parts of Pakistan, and virtually all of Bangladesh. This back-trajectory cluster category represents air with very-likely influence of anthropogenic and other continental sources from Southern India and Sri Lanka. Atmospheric transport from the IGP, the BoB, and Southern India/Sri Lanka to MCOH belongs to this category, with typical transport times of 5 to 10 days until arrival at the site. The curved nature of the cluster trajectory through the Bay of Bengal is due to the persistent anticyclone circulation over the Indian subcontinent during the NE monsoon season.
Bay of Bengal (BoB) forms the northeastern part of the Indian Ocean. It is bordered mostly by India and Sri Lanka to the west, Bangladesh to the north, and Myanmar to the east. For the BoB-origin, air mass back trajectories mostly transect over the BoB and then through the southern part of India or Sri Lanka to reach MCOH.
Arabian Sea (AS) is located on the west side of the northern Indian Ocean, surrounded by Pakistan and Iran to the north and by NE Somalia and the Arabian Peninsula on the west, and by India on the east. Air masses of the AS-origin is particularly common at MCOH during the NE Monsoon and inter-monsoon periods.
3.2 Particulate mass and total particle number concentrations
Particle size is a fundamental property of an aerosol particle. In addition, the aerosol number, aerosol mass and chemical composition of the atmospheric aerosols are all contributors to the diverse processes that continuously determine how the ever-changing aerosol population evolve. These aerosol properties will be successively described for the northern Indian Ocean winter regime in the following sections starting with mass concentrations and total particle number concentrations.
The highest mean concentration of total mass in PM2.5 (23 ± 7 μg m−3) and in PM10 (33 ± 6 μg m−3) were observed at MCOH in the air masses of IGP-origin (Table and Fig. ). In contrast, the lowest mean values of PM2.5 (12 ± 6 μg m−3) and of PM10 (23 ± 7 μg m−3) mass concentration were seen when the air masses came from over AS. The current study establishes that the fine fraction measured in PM2.5 also made up an overwhelming part also of the total PM10 for all three air masses. On average, PM2.5 mass of PM10 accounted for ~54% in the AS-origin, ~61% in the BoB-origin and ~70% in the IGP-origin. This observation suggests that the proportions of PM2.5 contained within PM10 were quite high, compared to those documented over the Bay of Bengal by Budhavant et al. (Citation2010) while more similar to those reported by Bikkina et al. (Citation2011) (i.e., PM2.5 accounted for ~65% of PM10 over the BoB for the air masses from IGP-origin). The somewhat higher bulk mass for PM10 is likely related to sea salt aerosols. The partitioning into the fine PM2.5 fraction was greater for the carbonaceous components of aerosols as espoused below. This holds true also for non-sea-salt major ion components, as also shown below.
Table 1. Statistical details of aerosol number, aerosol mass, various ionic concentrations and CO measured at MCOH over the full winter period November 2014 to April 2015.
Fig. 2. Comparison of EC, OC, PM2.5 (mass) and a daily average of total particle number concentration measured at Maldives Climate Observatory-Hanimaadhoo in full winter seasons ten years apart. The 2004–05 data are from Ramanathan et al. (Citation2007a).
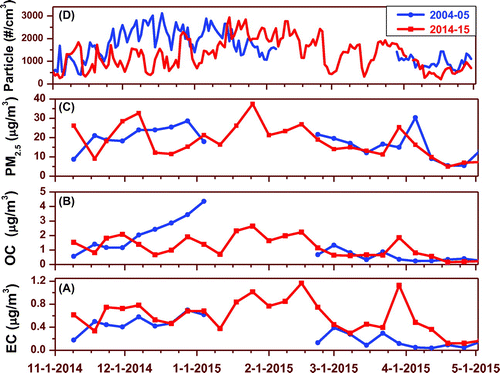
The atmospheric lifetime of fine mode aerosols in the IGP outflow is estimated to be about a week. This assessment is based on two independent approaches. First, the washout ratios of chemical constituents in the simultaneously collected rainwater and aerosol samples (Budhavant et al., Citation2010; Granat et al., Citation2010); and secondly, on the measured 210Pb activity in the PM2.5 from the IGP (Bikkina et al., Citation2015). The suggested 7–10 day timescale allows for long-range transport of a significant fraction of aerosols from as far as the N. India region to MCOH. However, coarse mode particles, mainly produced by mechanical processes from both natural and anthropogenic sources, are apparently scavenged before reaching MCOH. At MCOH, sea salt aerosols are therefore expected to be the dominant contributor to the coarse mode. A comparison of total aerosol mass concentrations from the same site as well as other locations in the S. Asian region is presented in Table . Across S. Asia, PM2.5 mass concentrations have been reported between 17 and 277 μg m−3 (Table ). In summary, the total particle mass was partitioned between both coarse and fine modes, with the coarse mode likely dominated by sea salt aerosols, and the fine mode likely hosting most of the anthropogenic aerosols.
Table 2. Aerosol mass and carbonaceous species (μg/m3) from different sites in South Asia and adjacent seas and regions.
The total particle number concentration measured with the CPC is shown in Fig. . During the measurement period, the daily particle number concentration varied between 200- 3000 cm−3 (av. 1300 ± 700 cm−3). Rapid increases in daily particle number concentration at MCOH were mainly associated with air masses carrying continental pollutants from the IGP region (1700 ± 650 cm−3). The daily average aerosol number concentrations for the BoB and AS aerosols were 1500 ± 450 and 650 ± 330 cm−3, respectively. The measured variability of aerosol mass and number concentration at MCOH thus clearly depends on meteorological conditions.
The aerosol number concentration was mostly affected by wind speed (r = -0.82) and to some extent by precipitation (or rainfall) events (Table S2). The rain plays an important role in modulating the aerosol size distribution as the rainfall more efficiently scavenges larger particles (Hyvärinen et al., Citation2011; Budhavant et al., Citation2012). Occasionally, very high particle number concentrations were found during midday hours. This is perhaps linked to new particle formation events over the ocean surface. The earliest reported aerosol number concentration data from MCOH (Ramanathan et al., Citation2007a) also show a similar kind of daily average variation (400–3100 cm−3) for the dry season 2004–2005 as shown in Fig. .
3.3 Aerosol number-size distributions
Particle number size distribution and its variations were explored to investigate sources/formation and properties of the aerosol regime. During the Indian Ocean Experiment (INDOEX), the majority of detected particles with aerodynamic diameters between 0.3 and 1.0 μm were classified as carbon-containing particles by an aerosol time-of-flight mass spectrometer (Guazzotti et al., Citation2003). To explore the size distribution also of sub-micron particles (d < 1 μm), we measured aerosol size distributions in the range 0.007–0.8 μm using the DMPS. Size distributions were plotted separately for the three previously described source regions and presented as an average for the entire study period (Fig. ). The averaged size distributions of all the three clusters showed a modal peak close to 0.09 μm, indicative of aged and processed aerosols. The tails of the size distributions into larger sizes is strongly suggestive of cloud processing. The bi-modal nature of the distributions, typical to marine cloud-processed aerosol, becomes even more evident in Figs. S4–S6. They also show that the Arabian Sea cluster is the most diverse and IGP cluster is the most homogeneous. The Arabian Sea cluster is characterized by large variability in both concentrations and size distributions (Fig. S6). In the IGP cluster (Fig. S4), the Hoppel minimum (Hoppel et al., Citation1986) in the size distribution (about 0.15–0.20 μm) is indicative of the minimum size of aerosols that are activated. This minimum is the result of a selective activation of larger aerosol particles in the cloud forming process. Upon cloud evaporation, larger aerosol particles that had been activated create an accumulation-size mode that is distinct from the smaller aerosol particles that have not increased in mass by aqueous phase chemistry. Aqueous phase chemistry, the oxidation of SO2 to sulphate adds solute mass. The bimodal distribution of the aged aerosol is therefore most likely the result of atmospheric processing of regional haze particles during several days.
Fig. 3. The average particle number size distributions using Differential Mobility Particle Sizer (DMPS), resulting from grouping the data into three source regions. The numbers in coloured boxes present the average total number concentration for each of the sources.
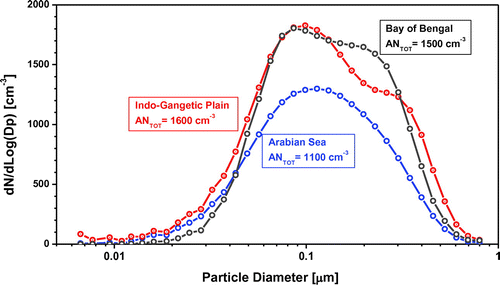
Most of the particle numbers found at MCOH during the study period are in the ‘accumulation mode’ (0.05 to 0.40 μm). These accumulation mode particles are removed from the atmosphere mainly by wet deposition. As aerosol particles in accumulation mode can remain in the troposphere for a long time (7–10 days) due to their slow removal, especially during dry season, they can be transported long distances from their sources. We have also used the HYSPLIT trajectory model to calculate rainfall to estimate and see wet deposition effects during transport. We found that the wet deposition effect was negligible, especially when trajectories come from the IGP (Figs. S7 and S8).
A unimodal size distribution was observed when the trajectories had not passed over land for the last 7–10 days. Accumulation mode particles over MCOH most likely originate from coagulation and condensational growth of Aitken mode particles advected to the site through long-range transport from South Asia. In addition, cloud processing is also likely to have grown particles into the Accumulation mode. Previous research in this region shows that the low frequency of rainfall during the wintertime Indian NE monsoon period allows for the build-up of aerosol pollution due to strong sources and limited advection (e.g., Budhavant et al., Citation2015a), which subsequently can be observed as a brown cloud over extensive spatial scales of South Asia and N. Indian Ocean (e.g., Ramanathan et al., Citation2001; Corrigan et al., Citation2006; Gustafsson and Ramanathan, Citation2016).
3.4 Aerosol optical depth (AOD)
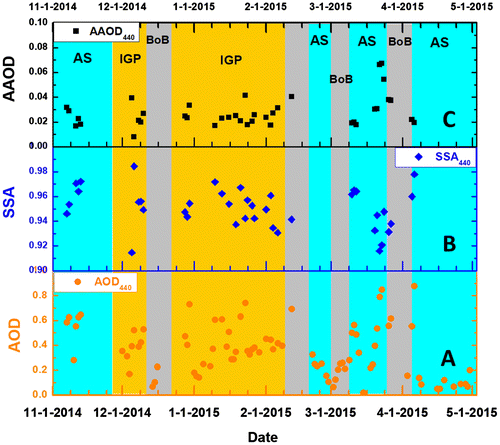
The AOD gives a columnar integrated picture of aerosol abundance at the MCOH sampling location. The AOD at MCOH varied between 0.1 and 0.8 with an average of 0.36 ± 0.22 for this period. The increase in columnar AOD and decrease in SSA were attributed to the arrival of air masses carrying more continental pollutants from IGP and the western margin of India that were characterized by relatively high concentrations of submicron absorbing aerosols (Fig. ). In INDOEX aircraft experiment data (Ramanathan et al., Citation2001; Citation2007b), most of the contribution to the increase in AOD comes from levels below 3 km. During the sampling period, columnar AAOD varies from 0.01 to 0.07 with an average of 0.03 (Fig. ). The highest values in AAODs normally match those regions that are heavily influenced by either regional forest fires or enhanced use of biomass fuels in densely populated developing countries (Laskin et al., Citation2015). This also supports that South Asia-IGP is a key regional hot spot source region defined by the criteria that anthropogenic AODs should exceed 0.3 and AAOD > 0.03 (Ramanathan et al., Citation2007a; Laskin et al., Citation2015). Experimental and modelling studies (e.g., Ramanathan et al., Citation2007b; Ten Hoeve et al., Citation2012; Lin et al., Citation2014) have recognized significant contributions from the Brown Carbon component (BrC, the light-absorbing organic carbon) to the observed AOD and AAOD values, and emphasized the critical need for a quantitative breakdown of the relative BrC and BC contributions as well as the need for advanced characterization of BrC, for improved climate predictions.
Fig. 4. Daily average aerosol optical depth (AOD), single-scattering albedo (SSA) and absorption aerosol optical depth (AAOD) measured at Maldives Climate Observatory-Hanimaadhoo using CIMEL Sun photometer during November 2014 to April 2016. Vertical colour shaded bars indicate the three different source regions: Arabian Sea (Cyan), Indo Gangetic Plain (Canary) and Bay of Bengal (Grey).
3.5 Aerosol chemical mass closure
The chemical mass closure was studied by comparing the mass derived from the chemical analysis to the gravimetric mass obtained by weighing the filter samples. PM mass closure applies multipliers to several of the measured species to estimate unmeasured components (e.g., Neususs et al., Citation2000; Chow et al., Citation2015). In this work, the mass of chemical components was calculated by summing up all measured ions, EC and organic matter (OM). Further, the chemical components were divided into four classes: secondary inorganic aerosols (nss-SO42–, NO3–, and NH4+) (SIA), sea salt, dust and carbonaceous aerosols (EC + OM). The mass closure requires a multiplication factor to bring OC measurements to estimates of the mass of the total particulate OM. Bikkina and Sarin, (Citation2014) recommended the conversion factor of 2.3 ± 0.6 for aged marine aerosols influenced by air masses from IGP and south-east Asia. In the present work a factor of 2.3 was chosen to estimate OM.
The contributions of the various components to the average gravimetric PM from different source regions were calculated and are shown in Fig. . The identified aerosol chemical components accounted for a higher fraction of the total PM for the IGP outflow (72% for PM10 and 65% in PM2.5) and for a lower fraction when the wind comes from the BoB (54% for PM10 and 38% in PM2.5). SIA and carbonaceous aerosols were predominantly found in PM2.5. On the other hand, sea salt and SIA were mainly present in PM10. Particularly nss-SO42− concentrations were significantly higher in total sulphate during IGP outflow (95% in PM2.5 and 82% for PM10) compared to in air masses coming from over the AS and BoB (Table ). The higher concentrations of nss-SO42− in the IGP outflow suggest a large input of non-biogenic sources. On the other side, the lower concentrations of nss-SO42− over the BoB and the AS representative of marine-biogenic sources.
Fig. 5. Mass closure of particulate matter in PM2.5 and PM10 measured at Maldives Climate Observatory-Hanimaadhoo from three different source regions: Indo Gangetic Plain (IGP), Bay of Bengal (BoB) and Arabian Sea (AS) during November 2014 to April 2015.
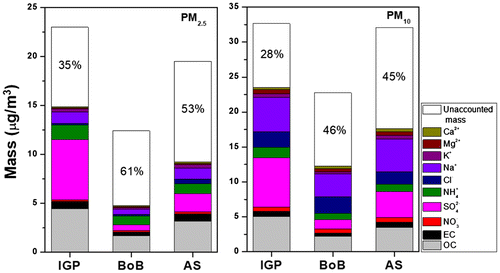
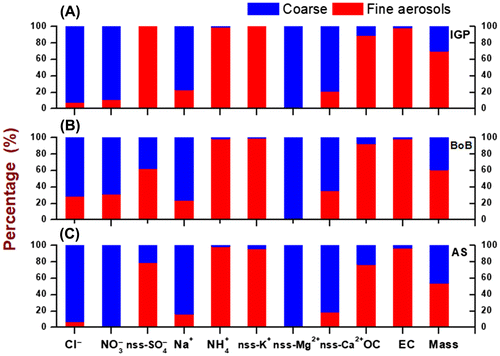
Fig. 6. Chemical composition and mass concentration of fine (PM2.5, red) and coarse (PM10–2.5 blue) aerosol from different source regions measured at MCOH. Panel A. Indo-Gangetic Plane (IGP), B. Bay of Bengal (BoB) and C. Arabian Sea (AS) during November 2014 to April 2015.
Likely explanations in the unaccounted mass in PM2.5 (54%) and PM10 (38%) include trace elements and aerosol-bound water. Another source of systematic error is the OC multiplication factor. In this study, the factor of 2.3 was applied for all the samples, although it is known that the particulate OC to OM ratio depends on the emission sources, atmospheric transport duration and meteorological conditions.
3.6 Characteristics of ionic aerosol components
The chemical characterization of aerosol particles is informative of their sources and transformation. PM2.5 and PM10 were characterized in terms of major cations (NH4+, Na+, K+, Mg2+ and Ca2+) and major anions (Cl−, NO3− and SO42−) for the six-month sampling period (Fig. ). Sulphate and ammonia dominate the PM2.5 mass, indicating strong anthropogenic sources for the ambient fine aerosol over the N. Indian Ocean. In the fine mode, SO42- (20%) showed the highest concentration followed by NH4+ (6%), Na+ (5%), K+ (2%), Cl− (1%), Mg2+ (1%); NO3− and Ca2+ were present in smaller amounts. In contrast, for the coarse fraction, Na+ (28%) showed the highest concentration followed by Cl− (17%), NO3− (4%), Mg2+ (3%), Ca2+ (2%), K+ (2%) and NH4+ (negligible amount). These observations clearly showed that marine components like Na+, Cl− and Mg2+ were dominant in coarse mode (48%) while their contribution was much less in the fine mode (7%). The major fraction of sea salt aerosols is generally found in the coarse size range (Heintzenberg et al., Citation1981; Porter and Clarke, Citation1997; Budhavant et al., Citation2016). The main source of sea salt aerosol over the open ocean is sea spray droplets (Blanchard and Woodcock, Citation1980; Martensson et al., Citation2003). In fact, the slope of the linear regression analysis of Na+ versus Mg2+ (R2 = 0.99; slope = 0.12) in PM10 is consistent with that typically observed for sea salt aerosols (Mg2+/Na+ ratio in Seawater: 0.12).
High concentrations of NO3− in the coarse mode at MCOH together with its clear linear relationship with Na+ in PM10 (R2 = 0.40; slope = 0.14) indicates the presence of NaNO3 as a result of the reactive uptake of gaseous phase nitric acid on sea salts. Over the ocean, the size distribution of NO3− normally follows the surface area distribution of sea salt (Li-Jones and Prospero, Citation1998; Murphy and Thomson, Citation1997). The composition was thus quite different for the fine fraction. The contribution of SO42−, NO3− and NH4+ together was much higher in the fine fraction (27%) than in the coarse fraction (5%). These are likely linked to long-range transport from anthropogenic emissions, originating primarily on the Indian subcontinent. By enlargement, it was thus found that anthropogenic components (nss-SO42−, nss-K+ and NH4+) were predominantly partitioned to the fine-sized aerosols, which was similar to what was also found for the carbonaceous components.
3.7 Characteristics of carbonaceous aerosol components
The distribution and properties of the carbonaceous aerosol fraction are important as they have proportionally a much larger impact on the aerosol absorption of solar radiation and thus on net radiative effects. EC normally makes up 3–10% of total aerosol mass in S. Asia (e.g., Lelieveld et al., Citation2001; Bikkina and Sarin, Citation2014; Budhavant et al., Citation2015a). The mean concentration of EC was found to be 0.59 ± 0.28 μg/m3 in PM2.5 and 0.60 ± 0.29 μg/m3 in PM10 (Table ). The highest EC concentrations were observed for the IGP-origin, 0.74 ± 0.17 μg/m3, whereas the lowest values were found for the Arabian Sea origin 0.38 ± 0.20 μg/m3 for PM2.5. The EC concentration measured during this study is almost double that previously reported at MCOH during the dry winter 2004–05, 0.27 μg/m3 (Fig. ). The temporal variations during winter 2014–2015 in concentrations of OC and EC (and the gaseous combustion marker CO) showed similar patterns (Fig. ).
Fig. 7. Time series of nss K+, EC and CO, as well as OC in PM2.5 and PM10 at Maldives Climate Observatory at Hanimaadhoo during November 2014 to April 2015. Vertical colour shaded bars indicate the three different source regions: Arabian Sea (Cyan), Indo Gangetic Plain (Canary) and Bay of Bengal (Grey).
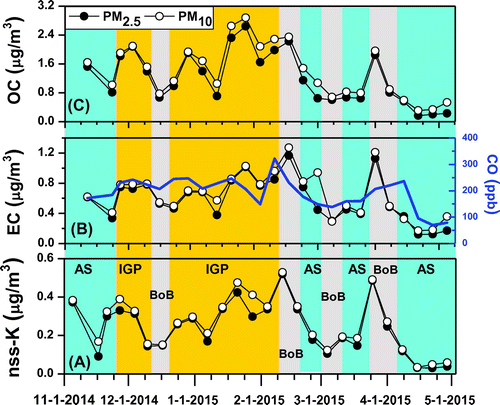
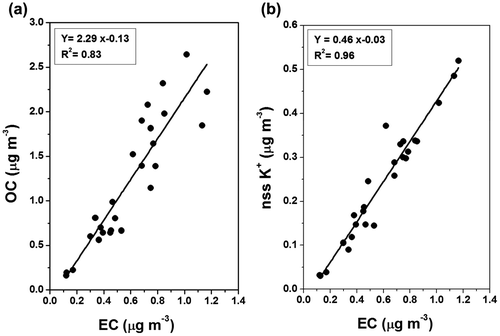
A significant correlation between OC and EC (slope = 2.28; R2 = 0.83 for PM2.5 and slope = 2.33; R2 = 0.81 for PM10; Fig. ) was observed during the study period, suggesting their common sources. EC is exclusively of primary origin as it is the product of incomplete combustion. Since EC undergoes minimal chemical transformation, it serves as a good tracer for anthropogenic aerosols. The co-variability of OC and EC together with their significant linear relationship and their near-exclusive partitioning to the fine PM2.5 fraction (Fig. ) suggests that the ambient concentration levels of not only EC but also of OC were controlled largely by primary emissions and atmospheric dispersion, rather than by any secondary OC formation. Table shows the concentrations of total aerosol mass as well as of various aerosol chemical components measured at MCOH in comparison to previous studies at different locations in mainland South Asia. Locations in IGP such as Lahore, Delhi, Kanpur and Dhaka are the most polluted (TC > 45 μg m−3). Lower but still high pollution (TC < 15 μg m−3) has been observed in cities like Karachi, Ahmedabad and Kanpur.
Fig. 8. Linear regression analysis between mass concentration of EC and (a) organic carbon, OC (b) non-sea-salt water-soluble potassium, (nss-K+) in PM2.5 collected over the MCOH during November 2014 – April 2015.
The contribution of total carbonaceous aerosols (TCA) to the total particulate mass concentration was also estimated. The TCA accounted for a higher fraction of total PM for the IGP outflow (24% in PM2.5 and 18% for PM10) with lower values when the wind comes over the BoB (16% PM2.5 and 11% for PM10). The average TCA was 20 ± 6% of PM2.5 and 14 ± 5% of PM10 at MCOH. These results are consistent with earlier reported values from the same station for PM2.5 (Ramanathan et al., Citation2007a; Budhavant et al., Citation2015a). Bikkina and Sarin (Citation2014) also reported similar TCA concentrations for the IGP source region (22 ± 6%, in PM2.5, 16 ± 3% in PM10).
3.8 Source categorization of chemical components of aerosols over northern Indian Ocean
Pinpointing the sources of carbonaceous aerosols is important in order to understand their atmospheric concentrations and processing, as well as to study their possible effects on climate and air quality. The mass ratio of OC to EC reflects multiple processes in the atmosphere (e.g., Kroll et al., Citation2011). The OC/EC ratios at MCOH ranged from 1.3 to 2.9 (avg. 2.0 ± 0.5) for PM2.5. The highest values were observed for IGP-origin, 2.9 (avg. 2.3) for PM2.5, whereas the lowest values were found for BoB-origin 1.3 (avg. 1.7) for PM2.5. These OC/EC ratios are quite low in comparison with other reported studies based in mainland South Asia except some urban cities like Dhaka and Chennai (Table ). Consistently low OC/EC ratios (2.0 ± 0.5), strong linear relationships between EC and OC, as well as the steady bimodal fine number-size distribution, combine to suggest a predominance of a common source for EC and OC.
We observed a significant linear relationship of not only EC with OC but also between EC and nss-K+ in the PM2.5 sampled at MCOH during November 2014–April 2015. The nss-K+ is a diagnostic proxy for identifying regional impact of biomass burning emissions (Andreae, Citation1983; Paris et al., Citation2010). Therefore, the former linear relationship indicates common source contributions and subsequent long-range atmospheric transport of carbonaceous species to MCOH, while the latter relationship emphasizes the significance of a biomass combustion source for EC and OC.
The MODIS active fire counts’ data (Fig. S9) showed high incidences of fires over India and Bangladesh during Nov 2014 to April 2015, also suggesting high emissions of biomass burning aerosols. The major contributions of carbonaceous aerosols reported from S Asia in cities like Kanpur (Ram et al., Citation2014), Ahmedabad (Rengarajan et al., Citation2011) and Karachi (Shahid et al., Citation2016) have been suggested to stem from biomass burning. At the same time, fossil fuel combustion has been suggested to be the major source of EC and OC in mega-cities such as Lahore (Alam et al., Citation2014), Delhi (Saxena et al., Citation2017), Dhaka (Begum et al., Citation2012) and Chennai (Pavuluri et al., Citation2011). Research using source-diagnostic 14C fingerprinting of ambient EC shows that fossil and biomass combustion processes are about equally responsible for the emission of EC over South Asia (Gustafsson et al., Citation2009; Budhavant et al., Citation2015b).
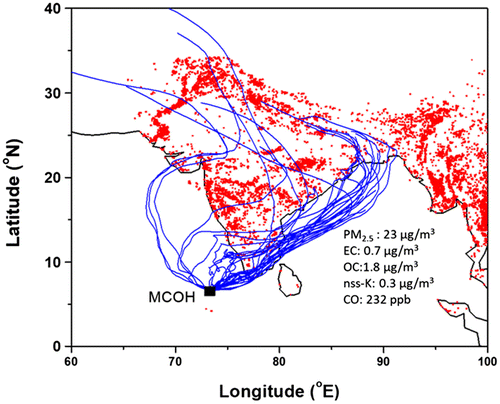
Based on the Peking University emission inventory (Wang et al., Citation2014), using a box [South: long. 70° to 90°; lat 5° to 15°, North: long 70° to 90°; lat 20° to 30°], shows that the emissions from the northern India is ~5 times higher than for the southern part. However, dispersion and atmospheric lifetimes affect the concentrations observed at MCOH. As aerosols and air masses arriving at MCOH is predominantly of IGP origin, it is likely that the wood /crop-residue burning emissions over the Indo-Gangetic Plain could be a major source of carbonaceous aerosols in the atmosphere over the N. Indian Ocean (Fig. ). This is further consistent with earlier studies of the diagnostic mass ratios of chemical constituents, organic molecular markers as well as radiocarbon source apportionment of aerosols that combine to suggest that postharvest crop residue and wood burning emissions contribute significantly to the S. Asian outflow (Ramanathan et al., Citation2007a; Rengarajan et al., Citation2007; Gustafsson et al., Citation2009; Bikkina and Sarin, Citation2014; Kirillova et al., Citation2014; Budhavant et al., Citation2015b).
Fig. 9. Air mass trajectories for 7 days ending at MCOH at 100 m with MODIS /Aqua active fires during the Indo-Gangetic Plain (IGP) outflow.
4. Conclusions
The aerosol regime over the N. Indian Ocean during the November 2014–April 2015 period is largely influenced by South Asian pollution. The chemical and physical properties of the air masses transported from the dominant IGP, Bay of Bengal and Arabian Sea source regions showed remarkably little variation during the six-month period of intensive observations at the Maldives Climate Observatory – Hanimaadhoo (MCOH).
The coarse mode was influenced by both sea salt aerosols and long-range transported aerosols of largely anthropogenic influence. In contrast the much more abundant (in mass and number) fine (PM2.5) aerosols were overwhelmingly dominated by anthropogenic particles, as indicated by the fingerprints of OC, EC, nss-SO42-, nss-K+, and NH4+. The EC, as a tracer of incomplete combustion, was 97 ± 3% in the fine mode.
Consistently low OC/EC ratios (2.0 ± 0.5), strong linear relationships between EC and OC as well as between nss-K+ (a proxy for biomass combustion) and both OC and EC, as well as the steady bimodal fine number-size distribution, combine to suggest a predominance of a common primary source, with a large biomass burning contribution.
Taken all together, the wintertime high-AOD, high-AAOD aerosol regime over N. Indian Ocean appears to be well homogenized, and dominated by the fine fraction for key components such as EC and OC, which in turn appear to be predominantly of primary origin with biomass burning as a putatively major source. These results for the sparsely studied, but high-AOD, high-AAOD regime of the N. Indian Ocean facilitates our understanding of the sources and properties of its aerosol regime.
Supplementary material
Supplemental data for this article can be accessed here 10.1080/16000889.2018.1464871.
Funding
This work was financially supported for this investigation from the Swedish funding agencies FORMAS (Grant 942-2015-1061), the Swedish Energy Agency (STEM, 2015-002538), and the Swedish Research Council (VR Grants 2015-03279 and 2017-01601). One of the authors (KB) thank the additional support from the VINNMER Marie Curie Programme (2016-04078) and Divecha Centre for Climate Change, Indian Institute of Science for their support. The Finnish Meteorological Institute research was supported by the Academy of Finland Center of Excellence programme (grant number 307331) and the Academy of Finland project ICASIF (grant number 284536).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary Material
Download MS Word (4.1 MB)Acknowledgements
The authors are grateful to the Maldives Meteorological Service and Ministry of Environment, Republic of the Maldives, for their cooperation and encouragement. The MCOH technicians Mr. Sharafulla Thoha and Miss. Mariyam provided assistance during the campaign. The authors are also thankful to Prof. V. Ramanathan and Dr P.S. Praveen for providing easy-access to published data-sets for comparison.
Related Research Data
References
- Alam, K., Mukhtar, A., Shahid, I., Blaschke, T., Majid, H. and co-authors 2014. Source apportionment and characterization of particulate matter (PM10) in urban environment of Lahore. Aerosol Air Qual. Res. 14, 1851–1861.
- Andreae, M. O. 1983. Soot carbon and excess fine potassium: long-range transport of combustion-derived aerosols. Science 220(4602), 1148–1151.10.1126/science.220.4602.1148
- Begum, B. A., Hossain, A., Nahar, N., Markwitz, A. and Hopke, P. K. 2012. Organic and black carbon in PM2.5 at an urban site at Dhaka, Bangladesh. Aerosol. Air Qual. Res. 12, 1062–1072.
- Bikkina, S. and Sarin, M. M. 2014. PM2.5, EC and OC in atmospheric outflow from the Indo-Gangetic Plain: Temporal variability and aerosol organic carbon-to-organic mass conversion factor. Sci. Tot. Environ. 487, 196–205.
- Bikkina, S., Andersson, A., Sarin, M. M., Sheesley, R. J., Kirillova, E. and co-authors, 2016. Dual carbon isotope characterization of total organic carbon in wintertime carbonaceous aerosols from northern India. J. Geophys. Res. Atmos. 121, 4797–4809. doi:10.1002/2016JD024880.
- Bikkina, S., Sarin, M. M. and Chinni, V. 2015. Atmospheric 210 Pb and anthropogenic trace metals in the continental outflow to the Bay of Bengal. Atmos. Environ. 122, 737–747.10.1016/j.atmosenv.2015.10.044
- Bikkina, S., Sarin, M. M. and Sarma, V. V. S. S. 2011. Atmospheric dry deposition of inorganic and organic nitrogen to the Bay of Bengal: Impact of continental outflow. Mar. Chem. 127(1–4), 170–179.
- Birch, M. E. and Cary, R. A. 1996. Elemental carbon-based method for monitoring occupational exposures to particulate diesel exhaust. Aerosol Sci. Technol. 25(3), 221–241.10.1080/02786829608965393
- Blanchard, D. C. and Woodcock, A. H. 1980. The production, concentration, and vertical distribution of the sea-salt aerosol. Ann. N. Y. Acad. Sci. 338, 330–347.10.1111/nyas.1980.338.issue-1
- Bond, T. C., Doherty, S. J., Fahey, D. W., Forster, P. M., Berntsen, T. and co-authors 2013. Bounding the role of black carbon in the climate system: a scientific assessment. J. Geophys. Res. Atmos. 118, 5380–5552.10.1002/jgrd.50171
- Bosch, C., Andersson, A., Kirillova, E. N., Budhavant, K., Tiwari, S. and co-authors 2014. Source-diagnostic dual-isotope composition and optical properties of water-soluble organic carbon and elemental carbon in the South Asian outflow intercepted over the Indian Ocean. J. Geophys. Res. Atmos. 119, 11,743–11,759. DOI:10.1002/2014JD022127.
- Boucher, O., Randall, D., Artaxo, P., Bretherton, C., Feingold, G. and co-authors. 2013. Clouds and aerosols. In: Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change (ed. T. F. Stocker, D. Qin, G.-K. Plattner, M. Tignor, S. K. Allen and co-authors. Cambridge University Press, Cambridge, UK, and New York, NY, USA.
- Budhavant, K. B., Rao, P. S. P., Safai, P. D., Gawhane, R. D., Raju, M. P. and co-authors 2012. Atmospheric wet and dry depositions of ions over an urban location in South-West India. Aerosol Air Qual. Res. 12, 561–570.
- Budhavant, K. B., Rao, P. S. P., Safai, P. D., Gawhane, R. D. and Raju, M. P. 2010. Chemistry of rainwater and aerosols over Bay of Bengal during CTCZ program. J. Atmos. Chem. 65, 171–183.10.1007/s10874-011-9187-0
- Budhavant, K. B., Rao, P. S. P. and Safai, P. D. 2016. Size distribution and chemical composition of summer aerosols over southern ocean and the Antarctic region. J. Atnos. Chem.. doi:10.1007/s10874-016-9356-2.
- Budhavant, K., Andersson, A., Bosch, C., Krusa, M., Murthaza, A. and co-authors. 2015a. Apportioned contributions of PM2.5 fine aerosol particles over the Maldives (northern Indian Ocean) from local sources vs long-range transport. Sci. Total Environ. 536, 72–78. doi:10.1016/j.scitotenv.2015.07.059.
- Budhavant, K., Andersson, A., Bosch, C., Kruså, M., Kirillova, E. N. and co-authors. 2015b. Radiocarbon-based source apportionment of elemental carbon aerosols at two South Asian receptor observatories over a full annual cycle. Environ. Res. Lett. 10(6), 064004. doi:10.1088/1748-9326/10/6/064004.
- Chow, J. C., Lowenthal, D. H., Chen, L. W. A., Wang, X. and Watson, J. G. 2015. Mass reconstruction methods for PM2.5: a review. Air Qual Atmos Health. 8, 243–263. doi:10.1007/s11869-015-0338-3. 10.1007/s11869-015-0338-3
- Corrigan, C. E., Ramanathan, V. and Schauer, J. J. 2006. Impact of monsoon transitions on the physical and optical properties of aerosols. J. Geophys. Res. 111, 817.10.1029/2005JD006370
- Granat, L., Engström, J. E., Praveen, S. and Rodhe, H. 2010. Light absorbing material (soot) in rainwater and in aerosol particles in the Maldives. J. Geophys. Res. 115, D22S22.10.1029/2009JD013768
- Guazzotti, S. A., Suess, D. T., Coffee, K. R., Quinn, P. K., Bates, T. S. and co-authors. 2003. Characterization of carbonaceous aerosols outflow from India and Arabia: Biomass/biofuel burning and fossil fuel combustion. J. Geophys. Res. 108(D15), 211. DOI:10.1029/2002JD003277.
- Gustafsson, Ö and Ramanathan, V. 2016. Convergence on climate warming by black carbon aerosols. Proc. Natl. Acad. Sci. U S A. 113(16), 4243–4245. doi:10.1073/pnas.1603570113.
- Gustafsson, O., Krusa, M., Zencak, Z., Sheesley, R. J., Granat, L. and co-authors. 2009. Brown clouds over South Asia: Biomass or fossil fuel combustion. Science 323(5913), 495–498. DOI:10.1126/science.1164857.
- Heintzenberg, J., Hansson, H. C. and Lannefors, H. 1981. The chemical composition of arctic haze at Ny-Alesund. Spitsbergen. Tellus 33(2), 162–171.
- Holben, B. N., Eck, T. F., Slutsker, I., Tanre, D., Buis, J. P. and co-authors. 1998. AERONET – A federated instrument network and data archive for aerosol characterization. Remote. Sens. Environ. 66, 1–16.
- Holben, B. N., Tanré, D., Smirnov, A., Eck, T. F., Slutsker, I., Abuhassan, N. and co-authors. 2001. An emerging ground-based aerosol climatology: aerosol optical depth from AERONET. J. Geophys. Res. 106, 12067–12097.10.1029/2001JD900014
- Hoppel, W. A., Frick, G. M. and Larson, R. E. 1986. Effect of nonprecipitating clouds on the aerosol size distribution in the marine boundary layer. Geophys. Res. Lett. 13, 125–128.10.1029/GL013i002p00125
- Hyvärinen, A. P., Raatikainen, T., Komppula, M., Mielonen, T., Sundström, A. M. and co-authors. 2011. Effect of the summer monsoon on aerosols at two measurement stations in Northern India – Part 2: Physical and optical properties. Atmos. Chem. Phys. 11, 8283–8294.10.5194/acp-11-8283-2011
- Janssen, N. A. H., Gerlofs-Nijland, M. E., Lanki, T., Salonen, R. O., Cassee, F. and co-authors. 2012. Health Effects of Black Carbon. The WHO 30 European Centre for Environment and Health, Bonn. World Health Organisation Regional Office for Europe, Copenhagen, Denmark.
- Kanamitsu, M. 1989. Description of the NMC global data assimilation and forecast system. Wea. Forecasting. 4, 335–342.10.1175/1520-0434(1989)004<0335:DOTNGD>2.0.CO;2
- Kaspari, S., Painter, T. H., Gysel, M. and Schwikowski, M. 2013. Seasonal and elevational variations of black carbon and dust in snow and ice in the Solu-Khumbu, Nepal and estimated radiative forcings. Atmos. Chem. Phys. Discuss. 13, 33491–33521.10.5194/acpd-13-33491-2013
- Keene, W. C., Psxenny, A. A. P., Galloway, J. N. and Hawley, M. E. 1986. Sea-salt corrections and interpretation of constituent ratios in marine precipitation. J. Geophys. Res. 91, 6647–6658.
- Kirillova, E. N., Andersson, A., Sheesley, R. J., Kruså, M., Praveen, P. and co-authors. 2013. 13C-and 14C-based study of sources and atmospheric processing of water-soluble organic carbon (WSOC) in South Asian aerosols. J. Geophys. Res. Atmos. 118, 614–626. DOI:10.1002/jgrd.50130.
- Kirillova, E. N., Andersson, A., Tiwari, S., Srivastava, A. K., Bisht, D. S. and Gustafsson, Ö. 2014. Water-soluble organic carbon aerosols during a full New Delhi winter: Isotope-based source apportionment and optical properties. J. Geophys. Res. Atmos. 119, 3476–3485. DOI:10.1002/2013JD020041.
- Kroll, J. H., Donahue, N., Jimenez, J. L., Kessler, S. H., Canagaratna, M. R. and co-authors. 2011. Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol. Nature Chem. 3, 133–139.10.1038/nchem.948
- Laskin, A., Laskin, J. and Nizkorodov, S. A. 2015. Chemistry of atmospheric brown carbon. Chem. Rev. 115, 4335–4382.10.1021/cr5006167
- Lawrence, M. and Lelieveld, J. 2010. Atmospheric pollutant outflow from southern Asia: a review. Atmos. Chem. Phys. 10(22), 11017–11096.10.5194/acp-10-11017-2010
- Lelieveld, J., Crutzen, P. J., Ramanathan, V., Andreae, M. O., Brenninkmeijer, C. A. M. and co-authors 2001. The Indian ocean experiment: widespread air pollution from South and Southeast Asia. Science 291(5506), 1031–1036.10.1126/science.1057103
- Li, C., Bosch, C., Kang, S., Andersson, A., Chen, P. and co-authors. 2016. Sources of black carbon to the Himalayan-Tibetan Plateau glaciers. Nat. Commun. 7(12), 574.
- Li-Jones, X. and Prospero, J. M. 1998. Variations in the size distribution of non-sea-salt sulfate aerosol in the marine boundary layer at Barbados: Impact of African dust. J. Geophys. Res. 103(D13), 16073–16084.10.1029/98JD00883
- Lin, G., Penner, J. E., Flanner, M. G., Sillman, S., Xu, L. and Zhou, C. 2014. Radiative forcing of organic aerosol in the atmosphere and on snow: Effects of SOA and brown carbon. J. Geophys. Res. 119, 7453.
- Martensson, E. M., Nilsson, E. D., de Leeuw, G., Cohen, L. H. and Hansson, H. C. 2003. Laboratory simulations and parameterization of the primary marine aerosol production. J. Geophys. Res. 108, 4297.
- Menon, S., Koch, D., Beig, G., Sahu, S., Fasullo, J. and Orlikowski, D. 2010. Black carbon aerosols and the third polar ice cap. Atmos. Chem. Phys. 10, 4559–4571.10.5194/acp-10-4559-2010
- Murphy, D. M. and Thomson, D. S. 1997. Chemical composition of single aerosol particles at Idaho Hill: negative ion measurements. J. Geophys. Res. 102, 6353–6368.10.1029/96JD00859
- Neusüss, C., Pelzing, M., Plewka, A. and Herrmann, H. 2000. A new analytical approach for size-resolved speciation of organic compounds in atmospheric aerosol particles: Methods and first results. J. Geophys. Res. 105, 4513–4527.10.1029/1999JD901038
- Pant, P., Shukla, A., Kohl, S. D., Chow, J. C., Watson, J. G. and Harrison, R. M. 2015. Characterization of ambient PM2.5 at a pollution hotspot in New Delhi, India and inference of sources. Atmos. Environ. 109, 178–189.10.1016/j.atmosenv.2015.02.074
- Paris, R., Desboeufs, K. V., Formenti, P., Nava, S. and Chou, C. 2010. Chemical characterisation of iron in dust and biomass burning aerosols during AMMA-SOP0/DABEX: implication for iron solubility. Atmos. Chem. Phys. 10, 4273–4282.10.5194/acp-10-4273-2010
- Pavuluri, C. M., Kawamura, K., Aggarwal, S. G. and Swaminathan, T. 2011. Characteristics, seasonality and sources of carbonaceous and ionic components in the tropical aerosols from Indian region. Atmos. Chem. Phys. 11, 8215–8230.10.5194/acp-11-8215-2011
- Porter, J. and Clarke, A. 1997. Aerosol size distribution models based on in situ measurements. J. Geophys. Res. 102, 6035–6045.10.1029/96JD03403
- Rahman, S. A., Hamzah, M. S., Elias, M. S., Salim, N. A. A., Hashim, A. and co-authors 2015. A long term study on characterization and source apportionment of particulate pollution in Klang Valley, Kuala Lumpur. Aerosol Air. Qual. Res. 15, 2291–2304.
- Raja, S., Biswas, K. F., Husain, L. and Hopke, P. K. 2009. Source apportionment of the atmospheric aerosol in Lahore, Pakistan. Water Air Soil Pollut. 208(1–4), 43–57.
- Ram, K., Sarin, M. M. and Tripathi, S. N. 2012. Temporal Trends in Atmospheric PM2.5, PM10, Elemental Carbon, Organic Carbon, Water-Soluble Organic Carbon, and Optical Properties: Impact of Biomass Burning Emissions in the Indo-Gangetic Plain. Environ. Sci. Technol. 46, 686–695.10.1021/es202857w
- Ram, K., Tripathi, S. N., Sarin, M. M. and Bhattu, D. 2014. Primary and secondary aerosols from an urban site (Kanpur) in the Indo-Gangetic Plain: impact on CCN, CN concentrations and optical properties. Atmos. Environ. 89, 655–663.10.1016/j.atmosenv.2014.02.009
- Ramanathan, V., Crutzen, P. J., Kiehl, J. T. and Rosenfeld, D. 2001. Atmosphere-Aerosols, climate, and the hydrological cycle. Science 294, 2119–2124.10.1126/science.1064034
- Ramanathan, V., Li, F., Ramana, M. V., Praveen, P. S., Kim, D. and co-authors. 2007a. Atmospheric brown clouds: Hemispherical and regional variations in long-range transport, absorption, and radiative forcing. J. Geophys. Res.: Atmos 112, D22S21.
- Ramanathan, V., Ramana, M. V., Roberts, G., Kim, D., Corrigan, C. and co-authors 2007b. Warming trends in Asia amplified by brown cloud solar absorption. Nature 448, 575–578.10.1038/nature06019
- Rengarajan, R., Sarin, M. and Sudheer, A. 2007. Carbonaceous and inorganic species in atmospheric aerosols during wintertime over urban and high-altitude sites in North India. J. Geophys. Res. 112, 32119. DOI:10.1029/2006JD008150.
- Rengarajan, R., Sudheer, A. K. and Sarin, M. M. 2011. Wintertime PM2.5 and PM10 carbonaceous and inorganic constituents from urban site in western India. Atmos. Res. 102, 420–431.10.1016/j.atmosres.2011.09.005
- Satheesh, S. K. and Ramanathan, V. 2000. Large differences in tropical aerosol forcing at the top of the atmosphere and Earth’s surface. Nature 405, 60–63.10.1038/35011039
- Saxena, M., Sharma, A., Sen, A., Saxena, P., Saraswati Mandal, T. K. and co-authors. 2017. Water soluble inorganic species of PM10 and PM2.5 at an urban site of Delhi, India: Seasonal variability and sources. Atmos. Res. 184, 112–125.10.1016/j.atmosres.2016.10.005
- Shahid, I., Kistler, M., Mukhtar, A., Ghauri, B. M., Ramirez-Santa Cruz, C. R.-S. and co-authors 2016. Chemical characterization and mass closure of PM10 and PM2.5 at an urban site in Karachi-Pakistan. Atmos. Environ. 128, 114–123.10.1016/j.atmosenv.2015.12.005
- Sheesley, R. J., Kirillova, E., Andersson, A., Kruså, M., Praveen, P. and co-authors 2012. Year-round radiocarbon-based source apportionment of carbonaceous aerosols at two background sites in South Asia. J. Geophys. Res. 117, D10202. DOI:10.1029/2011JD017161.
- Stein, A. F., Draxler, R. R., Rolph, G. D., Stunder, B. J. B. and Cohen, M. D. 2016. NOAA’s HYSPLIT Atmospheric Transport and Dispersion Modeling System. B. Am. Meteorol. Soc. 96, 2059–2077.
- Ten Hoeve, J. E., Jacobson, M. Z. and Remer, L. A. 2012. Comparing results from a physical model with satellite and in situ observations to determine whether biomass burning aerosols over the Amazon brighten or burn off clouds. J. Geophys. Res. 117, D08203.
- Wang, R., Tao, S., Balkanski, Y., Ciais, P., Boucher, O. and co-authors 2014. Exposure to ambient black carbon derived from a unique inventory and high-resolution model. Proc. Nat. Acad. Sci. 111, 2459–2463.10.1073/pnas.1318763111
- WHO. 2016. Country Estimates on Air Pollution Exposure and Health Impact. World Health Organisation. Online at: http://www.who.int/mediacentre/news/releases/2016/air-pollution-estimates/en/.