
ABSTRACT
Objectives: Beta-thalassemia (β-thal) is one of the most common genetic disorders worldwide. Multiple genetic and epigenetic mechanisms could be implicated in the pathogenesis and/or phenotype variations. We sought to explore the serum expression profile of three disease-related long non-coding RNAs (lncRNAs) in a sample of Egyptian β-thal patients with correlation to the patients’ clinicolaboratory data.
Methods: Fifty consecutive β-thal patients and 50 unrelated controls were enrolled in the study. Quantification of circulating lncRNAs; MALAT1 (metastasis-associated lung adenocarcinoma transcript 1), MIAT (myocardial infarction associated transcript), and ANRIL (antisense non-coding RNA in the INK4 locus) was done by Real-time qRT-PCR.
Results: Significant higher expression levels of the studied lncRNAs in β-thal patients compared to the controls (all P values < 0.001) were identified. There was no significant difference between β-thal-major and intermedia patients at the level of any of the studied lncRNAs. Higher MALAT1 expression profile was associated with early age at onset, early age at first blood transfusion, and a higher frequency of splenomegaly. Whereas, up-regulated MIAT levels were associated with early age at first blood transfusion.
Conclusions: Taken together, the studied lncRNAs MALAT1, MIAT, and ANRIL might be implicated in β-thal pathogenesis and could provide new molecular biomarkers for β-thalassemia after validation in large-scale future studies.
Introduction
β-thalassemia (β-thal) is a heterogeneous group of hemoglobin synthesis disorders, in which mutations reduce synthesis or stability of β-globin chain [Citation1]. The clinical and hematological types of β-thal over the past years were recognized as (1) β-thalassemia minor or carrier state results from heterozygous mutation of β-globin gene and is clinically asymptomatic or manifests as mild anemia and specific hematological characteristics, (2) β-thalassemia major (TM) is a severe transfusion-dependent anemia caused by homozygous or compound heterozygous mutation of β-globin gene and has several clinical manifestations, including facies, jaundice, hepatosplenomegaly, growth retardation, osteoporosis, and pathological fracture, and (3) β-thalassemia intermedia (TI) has clinical severity that lies between β-thal minor and TM [Citation2]. However, in 2012, the new terminology for β-thal clinical classification (i.e. transfusion-dependent thal and nontransfusion-dependent thal) was proposed and then adopted by the Thalassemia International Federation in their recent guidelines and publications [Citation3,Citation4].
β-thalassemia phenotypes cannot be solely explained by β-globin gene mutation because similar mutation occasionally produces a significantly distinct phenotype. In addition to β-globin gene mutations, hemoglobin peptide synthesis or balance could be influenced by other molecular modifiers including the epigenetic regulations [Citation2,Citation5]. The latter tend to be stochastic, reversible, and mosaic; their rules of occurrence and inheritance of epimutations are different from Mendelian genetics [Citation6]. One of the main players contributing to the epigenetic machinery is the long non-coding RNAs (lncRNAs) family [Citation7]. Increasing evidence showed that this type of non-coding RNAs (>200 nucleotide) could participate in various biological processes in health and disease, such as X-chromosome inactivation, genomic imprinting, quiescence, stress, immune response, cell differentiation, and several cellular developmental processes [Citation8]. Additionally, it has been reported to be implicated in hematopoiesis and the pathogenesis of blood diseases [Citation9–12].
Recently, Lai et al. have emphasized the contribution of lncRNAs to increased HbF levels by activating HBE1 expression and the hematopoietic cell lineage-inducible molecules, and inhibiting the apoptosis-inducible factors [Citation13]. Ma et al., in addition, suggested the relation of lncRNas to β-thalassemia clinical phenotype via their implications with their target genes in several phenotype-related pathways, such as response to hypoxia and muscle organ development, contributing to emergence of histanoxia, which is caused by elevated HbF levels and dysontogenesis in TM patients [Citation2].
As increased tissue hypoxia, oxidative stress, DNA damage and the release of microprticles have been related to many aspects of β thalassemia and its complications by several researchers especially those related to our region [Citation14,Citation15], we were interested to investigate three disease-related lncRNAs which could be implicated in the occurrence of these events.
MALAT1 (metastasis-associated lung adenocarcinoma transcript 1), also named nuclear-enriched abundant transcript 2, encoded by MALAT1 gene on chromosome 11q13.1 with two exons (https://www.ncbi.nlm.nih.gov/gene/378938), is an ubiquitously expressed lncRNA in many normal cells and tissues [Citation16], initially associated to the severity of lung metastasis, but this observation has since been extended to many other diseases [Citation17]. It has been reported to be implied in alternative splicing regulation, interacting with SR (serien/arginine) proteins and splicing factors in nuclear speckle domains and it may act as a transcriptional regulator for numerous genes [Citation16,Citation18].
MIAT (myocardial infarction associated transcript) is another well-studied lncRNA [Citation19], encoded by MIAT gene, located on chromosome 22q12.1 with 5 exons (https://www.ncbi.nlm.nih.gov/gene/440823). At the molecular level, MIAT has a unique nuclear structure by evading nuclear export. Because of its physical interaction with the splicing factor 1 (SF1) via SF1- binding motif, it was speculated to be involved in RNA splicing and to exert a regulatory effect on gene expression [Citation20]. Furthermore, it was supposed to function as a competing endogenous RNA (ceRNA) to regulate the VEGF (vascular endothelial growth factor) levels by sponging the microRNA miR-150-5p, creating MIAT/miR-150-5p/VEGF loop that was implicated in microvascular dysfunction and other disorders [Citation21,Citation22].
The last lncRNA arranged to be investigated in the current study is ANRIL (antisense noncoding RNA in the INK locus), encoded by ANRIL gene located within the CDKN2B-CDKN2A gene cluster at chromosome 9p21 with 21 exons (https://www.ncbi.nlm.nih.gov/gene/100048912), is transcribed in the antisense direction to the INK4B-ARF-INK4A locus and is upregulated by the transcription factor E2F1 in an ATM-dependent manner after DNA damage [Citation23,Citation24]. Additionally, it can interact with the polycomb repressive complex-1 (PRC1) and -2 (PRC2), leading to epigenetic silencing of other genes in its cluster.
As no previous studies heitherto have reported on the above-mentioned lncRNAs expression in β-thalassemia, we aimed as a preliminary study to investigate for the first time the possible association of the circulating lncRNAs MALAT1, MIAT and ANRIL with β-thal in Egyptian patients and to correlate their relative expressions with the available patients’ laboratory and clinical data.
Subjects and methods
Study population
The present work is a case–control study conducted on beta-thalassemia patients attending the Hematology Pediatric Unit, Sohag University Hospital, Sohag, Egypt, during the period between June 2017 and January 2018. The study population consisted of 50 consecutive patients of both sexes with confirmed diagnosis of beta thalassemia and 50 controls. Differentiation between types of beta thalassemia was carried out according to the clinical severity and the blood transfusion requirments into blood transfusion-dependent thalassemia and blood transfusion-independent thalassemia which will be referred to thalassemia major (TM) and thalassemia intermediate, respectively in the current study [Citation25]. TM patients were on regular transfusion according to their age and body weight. Exclusion criteria included presence of acute infection at the time of blood collection, and the clinical and/or laboratory diagnosis of other types of anemia and other hemoglobinopathies. Controls were age- and sex-matched healthy children who accompanied their sibs attending the Pediatrics Clinic and had no previous history of blood transfusion for any reason during the preceding three months and have a negative family history for any hemoglobinopathies. Hemoglobin level measurements and hemoglobin electrophoresis were done for controls to exclude any blood-related disorders. The current study was approved by Ethics Review Committee, Faculty of Medicine, Sohag University. A written consent was obtained from the guardians of the patients before participation.
Clinical assessment of β-thalassemia patients
A structured interview form was used by the interviewer to collect data information about the patients’ medical history at their follow up visit in the Pediatrics clinics or during their stay in the hospital for regular blood transfusion. A pilot study was carried out, in which we pre-tested the designed questionnaire on five patients to ensure its feasibility.
Child growth assessment
The following growth indicators were used: (a) height-for-age to identify children who are stunted or tall, (b) weight-for-age to assess whether a child is underweight or severely underweight, (c) weight-for-height to identify wasted children and those at risk of becoming overweight or obese, and (d) Body Mass Index (BMI)-for-age for screening the overweight and obesity. Growth charts specific for Egyptian children were used [data source: Diabetes Endocrine Metabolism Pediatric Unit of Cairo University Children's Hospital]. The z-scores were then estimated by applying standard equations provided by WHO [Citation26]. Delayed puberty was diagnosed in boys and girls by absence of testicular development by age of 14 years and the absence of breast development by the age of 13 years respectively [Citation27,Citation28].
Hematological assessment
The pretransfusion hemoglobin level was done on cell Dyne-3700 fully automated cell counter (Abbott Diagnostics, Wiesbaden, Germany). The pretransfusion levels over one year period were traced from the past medical records of the patients and the average was considered as the mean pretransfusion hemoglobin level [Citation29]. The diagnosis of β-thal was confirmed by high-performance liquid exchange chromatography (HPLC) using Hemoglobin A1c/A2/F BIO-RAD D-10 dual program on D-10 analyzer (Bio-Rad, Gladesville, Australia). The hemoglobin fractions separation was based on its ionic interactions with the cartridge and its recorded absorbance at 415 nm after elution. The D-10 software performs reduction of the raw data and a chromatogram was generated which will have the peaks in the following order: Hb A1a, Hb A1b, Hb F, LA1c/CHb-1, Hb A1c, Hb P3, Hb A0, and Hb A2. The variant hemoglobin was identified by its retention times and comparing it with manufacturer assigned retention times. All the quality control measurements were run according to the manufacturer's instructions [Citation30].
LncRNAs MALAT1, MIAT and ANRIL expression profiling
The total RNA was extracted from the serum of all participants using Qiagen RNeasy Kit (Cat. No. 74104, Qiagen, Hilden, Germany) following the protocol supplied by the manufacturer. The potential genomic DNA contamination was eliminated by application of the DNase step in the provided protocol. The concentration and purity of the extracted total RNA at the absorbance ratio 260/280 nm were determined by NanoDrop ND-1000 spectrophotometer (NanoDrop Tech., Inc. Wilmington, DE, USA) and the integrity was checked on 2% agarose gel. Then high Capacity cDNA Reverse Transcription (RT) Kit (Applied Biosystems, P/N 4368814) was used for RT reaction as described in details in our previous work [Citation31]. The housekeeping gene GAPDH (Glyceraldehyde 3-phosphate dehydrogenase) was used for data normalization, and appropriate negative controls; a no template control (NTC) which omits RNA template from the reaction to test the possibility of extraneous nucleic acid contamination and a no reverse transcriptase control (NRT) which assesses the amount of DNA contamination (if any) present in an RNA preparation, were included in each run. The real-time PCR reactions for the three ncRNAs were carried out using specific Taqman probes (Hs00273907, Hs00402814, and Hs04406279 for MALAT1, MIAT and ANRIL, repectively) in a final volume of 20 µl which included 1.33 µl RT products, 10 µl 2× TaqMan Universal PCR Master Mix, 1 µl TaqMan RNA assay. The PCR was performed in StepOne™ Real-Time PCR System (Applied Biosystem, Foster City, California, United States) and incubated as follows: 95°C for 10 min followed by 40 cycles of 92°C for 15 s and 60°C for 1 min. All tests were performed in duplicate and the real-time PCR reactions were carried out in accordance with the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) guidelines [Citation32,Citation33].
Statistical analysis
Statistical Package for Social Science (SPSS) software version 23.0 was used for the statistical analyses. Sample size and power calculations using G power-3 software showed that with the specified study design (i.e. gene expression), allowable error rates; alpha error = 0.05, a medium effect size = 0.5, and sample size 50 for each group can give 80% power of the study (http://www.gpower.hhu.de/). Continuous data were presented as mean ± standard deviation (SD) or median (interquartile ranges) according to data distribution and variance homogeneity that have been checked by Shapiro–Wilk test and Levene test, respectively. Categorical variables were presented as frequency counts and compared using the chi-square (χ2) test, while student's t tests, Mann–Whitney U (MW) and Kruskal–Wallis (KW) tests used to compare continuous variables according to data type as mentioned. Non-parametric Spearman's rho correlation test was used to correlate serum lncRNAs expression levels. A two-tailed P < 0.05 has been considered statistically significant. The fold change of all lncRNAs expressions in each patient relative to the controls was calculated via Livak and Schmittgen [Citation34] method based on the quantitative cycle (Cq) value; 2−ΔΔCq.
Results
Baseline characteristics of the study population
Demographic and clinical characteristics of patients and controls are depicted in . No significant difference was found between the two groups. However, there was lower frequency of urban patients than rural residents (p = 0.022, odds ratio 0.40 and 95% confidence interval of 0.18–0.90). In the patient group, four fifth of patients had thalassemia major (TM) and only 20% had thalassemia intermedia (TI). TM patients had earlier age at onset (9.45 ± 6.51 months) compared to TI patients (27.4 ± 13.8 months), P = 0.002. Consistently, they started receiving blood transfusion at an earlier age (12.3 ± 12.2 months) than TI group (26 ± 6.9 months), P = 0.001. TM patients underwent higher rate of transfusion (P = 0.049) and more frequency of using iron chelators (P < 0.001). In addition, median levels of serum ferritin were1700ng/ml for TM and 600 ng/ml for TI (P = 0.013).
Table 1. Baseline characteristics of the study β-thalassemia patients (n = 50).
Child growth assessment
No significant difference between children in both patient and control groups were found regarding their growth assessment. However, TI patients had higher median height for age measurements compared to TM patients (P = 0.041) (Figure S1). Similarly, overall and stratified analysis by gender showed insignificant frequencies between groups regarding their height-for-age, weight-for-age, and BMI-for-age (Figure S2).
Hematological and molecular data
Overall analysis of thalassemia patients revealed mean values of HB electrophoresis (%) as following: HbA (21.1 ± 24.9), HbA2 (74.4 ± 26.4), and HbF (2.9 ± 0.53). No significant difference was encountered between TM and TI patient groups (Figure S3).
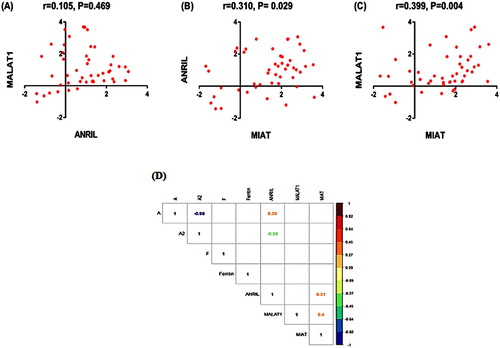
Molecular profiling of the three lncRNAs revealed significantly higher expression levels of ANRIL, MALAT1, and MIAT in thalassemia patients compared to control group (all P values < 0.001), . Median and interquartile range values of untransformed data were 11 (0.92–90) for ANRIL, 4.4 (1.8–69) for MALAT1, and 52 (1.8–291) for MIAT. There was no significant difference between β-TM and TI patients at the level of any of the studied lncRNAs. Correlation matrix demonstrated medium positive correlation between MIAT and both ANRIL (r = 0.310, P = 0.029) and MALAT1 (r = 0.399, P = 0.004) (). Regarding Hb electrophoresis, ANRIL expression levels showed moderate positive correlation to HbA and inversely correlation with HbA2 ((D)). Otherwise, none of the other study lncRNAs showed significant correlation with the Hb electrophoresis parameters.
Figure 1. Expression profile of lncRNAs in thalassemia patients and controls. Values are presented as medians. The box defines upper and lower quartiles (25% and 75%, respectively) and the whiskers bars indicate upper and lower adjacent limits. Fold-change (FC) for lncRNAs was normalized to GAPDH, calculated using delta-delta CT method [=2 (–ΔΔCT)] compared to controls, then log transformed. Mann–Whitney U test was used. aComparison to control group revealing statistical significance at P < 0.05. TM: thalassemia major, TI: thalassemia intermedia.
![Figure 1. Expression profile of lncRNAs in thalassemia patients and controls. Values are presented as medians. The box defines upper and lower quartiles (25% and 75%, respectively) and the whiskers bars indicate upper and lower adjacent limits. Fold-change (FC) for lncRNAs was normalized to GAPDH, calculated using delta-delta CT method [=2 (–ΔΔCT)] compared to controls, then log transformed. Mann–Whitney U test was used. aComparison to control group revealing statistical significance at P < 0.05. TM: thalassemia major, TI: thalassemia intermedia.](/cms/asset/adf34f5a-4b33-4139-b405-1644afde717e/yhem_a_1570616_f0001_ob.jpg)
Figure 2. Correlation analysis between lncRNA expression profiles (A–C). Correlation matrix (D) including the correlation between the study lncRNAs and the hematological laboratory parameters (only significant correlations are shown). Data are log transformed. Spearman's Rank correlation test was used.
Association of lncRNA expression levels with the clinical characteristics
presented the association between the three examined lncRNAs (ANRIL, MALAT1, and MIAT) and various clinical features of thalassemia patients. The higher MALAT1 profile was associated with early age at onset (P = 0.024), early age at first blood transfusion (P = 0.011), and higher frequency of splenomegaly (P = 0.014). Whereas, up-regulated MIAT levels were associated with early age at first blood transfusion (P = 0.008).
Table 2. Association between circulating lncRNAs relative expression levels and the clinical and laboratory data in β-thalassemia patients (n = 50).
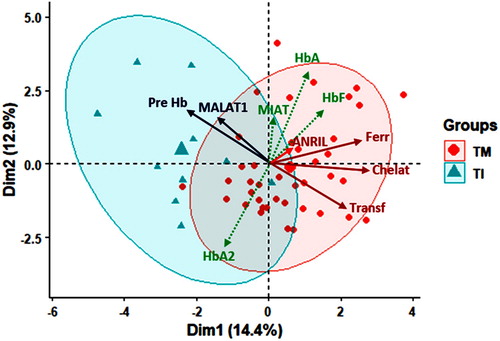
Multivariate analysis revealed partial clustering of patients with high ferritin, more frequent transfusion and chelation in thalassemia major group and higher MALAT1 level and pre-transfusion mean Hb level in intermedia patients ().
Figure 3. Principal component analysis for data exploration. Red circle surrounds the TM group while blue circle for TI group. Results were plotted on both axes. Red and blue solid lines influence TM and TI, respectively. Dim: dimension, TM: thalassemia major, TI: thalassemia intermedia, Pre-Hb: mean hemoglobin level pre-transfusion, HbA: adult Hb, HbF: fetal Hb, Ferr: ferritin, Chelat: chealtor use, Transf: blood transfusion.
Prognostic performance of lncRNAs expression
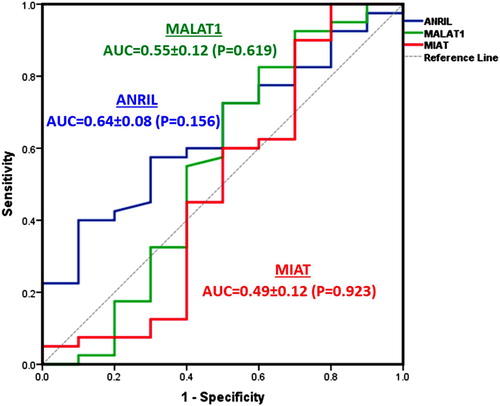
ROC analysis was applied to test the biomarker accuracy to differentiate between thalassemia major and intermedia (). Though all markers used were over-expressed in thalassemia diseases, but none of the lncRNAs exhibited high sensitivity or specificity to discriminate between the two types.
Figure 4. Diagnostic performance of the study lncRNAs expression to discriminate between thalassemia major and intermedia.
Discussion
Despite understanding the pathophysiology of thalassemia, particularly its molecular biology aspect, has improved over the past years, thalassemia-related outcomes still remain a significant problem carries a heavy social and economic burden. Recently, with high-throughput technology development, there are emerging evidences that suggest the important role of epigenetic mechanisms; including the non-coding RNAs, in thalassemia pathogenesis. These newly emerging RNAs can contribute to developing novel diagnostic and/or therapeutic strategies for β-thalassemia [Citation2].
In the present preliminary case–control study, for the first time we assessed the association of the expression profile of three speculated disease-related lncRNAs; MALAT1, MIAT, and ANRIL with β-thal and found that all the studied lncRNAs were significantly upregulated in β-thal patients compared with healthy controls. This could support their putative role in β-thal pathophysiology or progress.
The pathogenesis of many clinical manifestations pertained to thalassemia patients, especially those presented with TM phenotype, is multifactorial and one of these important factors is hypoxia [Citation35]. The latter could trigger a robust upregulation in MALAT1 expression in vitro and in vivo [Citation17,Citation36], as it has been found that MALAT1 gene has its own four HIF1α (hypoxia inducing factor 1 alpha)-binding sites corresponding to ‘the hypoxia response element ([A/G] CGTG), within 5.6 kb of the human MALAT1 promoter’ [Citation36]. Hence, through the enhanced activity of the CaMKK/AMPK/HIF-1α axis, it is likely that the increase in MALAT1 expression is part of an adaptive response to hypoxia [Citation17].
In addition, it has been found that MALAT1 depletion results in induction of the p53-mediated DNA damage response pathway in normal human diploid fibroblasts [Citation37]. This finding could add more insight to the speculated role of MALAT1 overexpression in thalassemia as an adaptive response to increased DNA damage, one of the major player in thalassemia phenotype and complications as mentioned above.
Given the diversity of the underlying molecular mechanisms implied by MALAT1, a recent high-throughput study identified many potential MALAT1-interacting proteins and established a highly connected MALAT1 interactome network [Citation38]. This latter study showed high involvement of MALAT1 in five biological processes: ‘RNA processing, gene transcription, ribosomal proteins, protein degradation, and metabolism regulation’ by gene ontology (GO) annotation and network analysis. These findings support the roles of the upregulated MALAT1 could play in β-thal which is characterized by various genetic and phenotypic features.
In an attempt to correlate serum levels of MALAT1 with the clinical and laboratory data of the patients, we found MALAT1 upregulation was significantly associated with early age at disease onset, early age at first blood transfusion, and higher frequency of splenomegaly which reflects higher rate of hemolysis that can result in lower baseline Hb levels [Citation39]. Both MALAT1 and the pre-transfusion mean hemoglobin level were the most prominent factors detected by the multivariate analysis influencing TI group. It has previously been observed that peripheral hemolysis contributing to anemia is less prominent in thalassemia major than in thalassemia intermedia, and occurs when insoluble alpha globin chains induce membrane damage to the peripheral erythrocytes [Citation40]. This observation could support the multivariate analysis findings. Interestingly, Danjou et al. highlighted the time to the first transfusion can accurately reflect variations in β-thalassemia phenotype severity regardless of patients’ major or intermedia phenotype based on blood transfusion frequency per year [Citation41]. This conclusion was supported by their findings that genetic modifiers including the different HbF production variants, α-globin gene defects, and unknown genetic factors, all had a substantial impact on the severity of β-thalassemia phenotype, allowing prediction of the need of patients to start transfusion at different times of their life [Citation42].
It is worth noting that the major players in β-thal pathophysiology are chronic anemia and hemolysis [Citation43] which result in persistent and severe hypoxia manifested by more severe β-thal phenotype in terms of the aforementioned manifestations. These observations support the association of MALAT1 expression with the severity of chronic hypoxia to which the tissues or cells exposed and suggests its implication in β-thal pathophysiology or may reflect more complex genetic and epigenetic molecular interactions at the cellular level that will need further exploration and validation in the future studies.
The lncRNA MIAT, also termed as Gomafu; the mouse homologue of MIAT, is an emerging functional player among all disease-related lncRNAs and is expressed in various tissues, including muscle, blood, nervous and retinal tissues, and exhibits deregulation in multiple diseases [Citation44]. Nearly no studies are available to date on the expression level of MIAT in peripheral blood from patients with thalassemia. By anology with hematopoietic cell lineage, Sattari et al., have reported upregulation of MIAT in lymphoid lineage with mature B phenotype including chronic lymphocytic leukemia and non-Hodgkin's lymphoma [Citation22]. Consistent with the regulatory loop MIAT constitute with OCT4 (octamer-binding transcription factor 4) in mouse embryonic stem cells [Citation45], the latter research group has shown a similar loop in lymphoma and has identified that both MIAT and OCT4 were essential for cell survival [Citation22]. Additionally, it has been reported that MIAT forms a feedback loop with vascular endothelial growth factor (VEGF) and microRNA miR-150-p to regulate endothelial cell function [Citation21] by functioning as a competing endogenous RNA (ceRNA). Clinical and experimental data suggest that endothelial dysfunction plays a pathogenic role in β-thal even in blood transfusion-independent model [Citation46,Citation47], and cardiac complications remain the main cause of mortality in both β-thalassemia major and intermedia [Citation48]. It can thus be hypothesized that increased MIAT expression in β-thal is associated with endothelial dysfunction. The finding of association of MIAT expression levels with early age at first blood transfusion suggests that its upregulated expression may be associated to the increased severity of β-thalassemia, a molecular finding which could be clinically implicated after its validation in larger cohorts.
Concerning the lncRNA ANRIL which is transcribed as an antisense RNA to INK4b, its elevated levels have been reported to suppress the expression of INK4A-ARF-INK4B locus in the late DNA damage response, an important mechanism to maintain genome integrity against intrinsic and extrinsic genotoxic stresses including reactive oxygen species generation, allowing negative feedback to this response [Citation23]. Interestingly, INK4b-ARF-INK4a locus was reported to be involved in hematopoietic differentiation, proliferation, and stress response [Citation49]. Thus, ANRIL helps the cell to return to a normal status at the completion of DNA repair [Citation20], supporting our current findings.
It is worth noting that the research pertaining to lncRNA in β-thalassemia is still in the early infancy, and some essential issues for its clinical application still need to be resolved. Their detailed regulatory mechanisms and their specified targets in β- thalassemia should be paid attention and validated before their final application in the clinic.
Conclusion
In summary, we showed for the first time that lncRNAs (MALAT1, MIAT, and ANRIL), are differentially expressed in β-thalassemia patients and healthy individuals which raise the possibility of their putative roles in β-thal pathophysiology. MALAT1 and MIAT, in addition, showed association with more severe phenotypic features. It is worth noting that although some of the current data were statistically insignificant, we acknowledge that the findings presented here are preliminary due to sample size limitation in some subgroups, as those stratified by type of thalassemia (TM vs. TI), which could be unable to explore the real difference. Thalassemia, in addition, is a complex disease, involving potential interactions among gene-gene and gene-environment. In this sense it is worth to explore the association of the study lncRNAs gene expression with different molecular defects of β-thalassemia in future studies either in the current population and/or others. Furthermore, larger and follow-up cohort studies are recommended to provide solid data to justify whether MALAT1 and MIAT could serve as new biomarkers for disease severity.
Acknowledgements
The authors thank all the participants for their approval to join this study.
Disclosure statement
No potential conflict of interest was reported by the authors.
ORCID
Abeer Fakhr-Eldeen http://orcid.org/0000-0002-2916-9542
Eman A. Toraih http://orcid.org/0000-0001-9267-3787
Manal S. Fawzy http://orcid.org/0000-0003-1252-8403
Related Research Data
References
- Thein SL. The molecular basis of β-thalassemia. Cold Spring Harb Perspect Med. 2013;3. doi:10.1101/cshperspect.a011700.
- Ma J, Liu F, Du X, et al. Changes in lncRNAs and related genes in β-thalassemia minor and β-thalassemia major. Front Med. 2017;11:74–86. doi:10.1007/s11684-017-0503-1.
- Cappellini MD, Cohen A, Porter J, et al. Guidelines for the Management of transfusion dependent Thalassaemia (TDT) [Internet]. 3rd ed. Nicosia Cyprus: Thalassaemia International Federation; 2014; Available from: https://www.ncbi.nlm.nih.gov/books/NBK269382/.
- Taher A, Vichinsky E, Musallam K, et al. Guidelines for the management of non transfusion dependent Thalassaemia (NTDT) [Internet]. Nicosia Cyprus: Thalassaemia International Federation; 2013; Available from: https://www.ncbi.nlm.nih.gov/books/NBK190453/.
- Kim A, Kiefer CM, Dean A. Distinctive signatures of histone methylation in transcribed coding and noncoding human β-globin sequences. Mol Cell Biol. 2007;27:1271–1279. doi:10.1128/MCB.01684-06.
- Martin DI, Ward R, Suter CM. Germline epimutation: a basis for epigenetic disease in humans. Ann N Y Acad Sci. 2005; 1054:68–77. doi: 10.1196/annals.1345.009
- Hanly DJ, Esteller M, Berdasco M. Interplay between long non-coding RNAs and epigenetic machinery: emerging targets in cancer? Philos Trans R Soc Lond B Biol Sci. 2018;373; doi:10.1098/rstb.2017.0074.
- Engreitz JM, Ollikainen N, Guttman M. Long non-coding RNAs: spatial amplifiers that control nuclear structure and gene expression. Nat Rev Mol Cell Biol. 2016;17:756–770. doi:10.1038/nrm.2016.126.
- Alvarez-Dominguez JR, Hu W, Yuan B, et al. Global discovery of erythroid long noncoding RNAs reveals novel regulators of red cell maturation. Blood. 2014;123:570–581. doi:10.1182/blood-2013-10-530683.
- Zhang X, Weissman SM, Newburger PE. Long intergenic non-coding RNA HOTAIRM1 regulates cell cycle progression during myeloid maturation in NB4 human promyelocytic leukemia cells. RNA Biol. 2014;11:777–787. doi:10.4161/rna.28828.
- Nobili L, Lionetti M, Neri A. Long non-coding RNAs in normal and malignant hematopoiesis. Oncotarget. 2016;7:50666–50681. doi:10.18632/oncotarget.9308.
- Kotani, A. Guest editorial: noncoding RNA in hematopoietic system. Int J Hematol. 2014;99:529. doi: 10.1007/s12185-014-1574-2
- Lai K, Jia S, Yu S, et al. Genome-wide analysis of aberrantly expressed lncRNAs and miRNAs with associated co-expression and ceRNA networks in β-thalassemia and hereditary persistence of fetal hemoglobin. Oncotarget. 2017;8:49931–49943. doi:10.18632/oncotarget.18263.
- Elsayh KI, Zahran AM, El-Abaseri TB, et al. Hypoxia biomarkers, oxidative stress, and circulating microparticles in pediatric patients with thalassemia in upper Egypt. Clin Appl Thromb Hemost. 2014;20:536–545. doi:10.1177/1076029612472552.
- Youssry I, Soliman N, Ghamrawy M, et al. Circulating microparticles and the risk of thromboembolic events in Egyptian beta thalassemia patients. Ann Hematol. 2017;96:597–603. doi:10.1007/s00277-017-2925-x.
- Zhang B, Arun G, Mao YS, et al. The lncRNA Malat1 is dispensable for mouse development but its transcription plays a cis-regulatory role in the adult. Cell Rep. 2012;2:111–123. doi:10.1016/j.celrep.2012.06.003.
- Sallé-Lefort S, Miard S, Nolin M, et al. Hypoxia upregulates Malat1 expression through a CaMKK/AMPK/HIF-1α axis. Int J Oncol. 2016;49:1731–1716. doi:10.3892/ijo.2016.3630.
- Tripathi V, Ellis JD, Shen Z, et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell. 2010;39:925–938. doi:10.1016/j.molcel.2010.08.011.
- Liao J, He Q, Li M, et al. LncRNA MIAT: myocardial infarction associated and more. Gene. 2016;578:158–161. doi:10.1016/j.gene.2015.12.032.
- Tsuiji H, Yoshimoto R, Hasegawa Y, et al. Competition between a noncoding exon and introns: Gomafu contains tandem UACUAAC repeats and associates with splicing factor-1. Genes Cells. 2011;16:479–490. doi:10.1111/j.1365-2443.2011.01502.x.
- Yan B, Yao J, Liu J-Y, et al. lncRNA-MIAT regulates microvascular dysfunction by functioning as a competing endogenous RNA. Circ Res. 2015;116:1143–1156. doi:10.1161/CIRCRESAHA.116.305510.
- Sun C, Huang L, Li Z, et al. Long non-coding RNA MIAT in development and disease: a new player in an old game. J Biomed Sci. 2018;25:23, doi:10.1186/s12929-018-0427-3.
- Wan G, Mathur R, Hu X, et al. Long non-coding RNA ANRIL (CDKN2B-AS) is induced by the ATM-E2F1 signaling pathway. Cell Signal. 2013;25:1086–1095. doi:10.1016/j.cellsig.2013.02.006.
- Kim C, Kang D, Lee EK, et al. Long noncoding RNAs and RNA-binding proteins in oxidative stress, cellular Senescence, and Age-related diseases. Oxid Med Cell Longev. 2017. doi: 10.1155/2017/2062384
- Viprakasit V, Ekwattanakit S. Clinical classification, screening and diagnosis for thalassemia. Hematol Oncol Clin North Am. 2018;32:193–211. doi:10.1016/j.hoc.2017.11.006.
- World Health Organization. Multicentre growth reference study group. WHO child growth standards: Length/height-for-age, weight-for-age, weight-for-length, weight-for-height and body mass index-for-age: Methods and development. 2006:312. Available at http://www.who.int/childgrowth/standards/technical_report/en/ (last accessed 24/7/2018).
- Pitteloud N. Managing delayed or altered puberty in boys. Br Med J. 2012;345; doi:10.1136/bmj.e7913.
- Sex steroid treatment for pubertal induction and replacement in the adolescent girl: Royal college of obstetricians and gynaecologists – scientific impact paper. 2013. Available at https://www.rcog.org.uk/en/guidelines-research-services/guidelines/sip40/ (last accessed 24/7/2018).
- Karunaratna AMDS, Ranasingha JGS, Mudiyanse RM. Iron overload in beta thalassemia major patients. Int J Blood Transfus Immunohematol 2017;7:33–40. doi: 10.5348/ijbti-2017-32-OA-5
- Chandrashekar V. Hb A1c separation by high performance liquid chromatography in hemoglobinopathies. Scientifica (Cairo). 2016. doi: 10.1155/2016/2698362
- Fawzy MS, Toraih EA, Abdallah HY. Long noncoding RNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1): A molecular Predictor of Poor survival in Glioblastoma Multiforme in Egyptian patients. Egypt J Medical Human Genet. 2017;18:1–239. doi:10.1016/j.ejmhg.2016.08.003.
- Bustin SA, Benes V, Garson JA, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55:11–622. doi:10.1373/clinchem.2008.112797.
- Bustin SA, Benes V, Garson J, et al. The need for transparency and good practices in the qPCR literature. Nat Methods. 2013;10:1063–1067. doi:10.1038/nmeth.2697.
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−△△CT method. Methods. 2001;25:402–408. doi:10.1006/meth.2001.1262.
- Kyriakou A. Skordis N. Mediterr J Hematol Infect Dis. 2009;1; doi:10.4084/MJHID.2009.003.
- Lelli A, Nolan KA, Santambrogio S, et al. Induction of long non coding RNA MALAT1 in hypoxic mice. Hypoxia (Auckl). 2015;3:45–52. doi:10.2147/HP.S90555.
- Tripathi V, Shen Z, Chakraborty A, et al. Long noncoding RNA MALAT1 controls cell cycle progression by regulating the expression of oncogenic transcription factor B-MYB. PLoS Genet. 2013;9; doi:10.1371/journal.pgen.1003368.
- Chen R, Liu Y, Zhuang H, et al. Quantitative proteomics reveals that long non-coding RNA MALAT1 interacts with DBC1 to regulate p53 acetylation. Nucleic Acids Res. 2017;45:9947–9959. doi:10.1093/nar/gkx600.
- Ekwattanakit S, Siritanaratkul N, Viprakasit V. A prospective analysis for prevalence of complications in Thai non-transfusion-dependent Hb E/β-thalassemia and α-thalassemia (Hb H disease). Am J Hematol. 2018;93:623–629. doi:10.1002/ajh.25046.
- Galanello R., Origa R. Beta-thalassemia. Orphanet J Rare Dis. 2010;5:11. doi: 10.1186/1750-1172-5-11
- Danjou F, Anni F, Perseu L, et al. Genetic modifiers of β-thalassemia and clinical severity as assessed by age at first transfusion. Haematologica. 2012;97:989–993. doi:10.3324/haematol.2011.053504.
- Danjou F, Francavilla M, Anni F, et al. A genetic score for the prediction of beta-thalassemia severity. Haematologica. 2015;100:452–457. doi:10.3324/haematol.2014.113886.
- Rund D, Beta-Thalassemia RE. N Engl J Med. 2005;353:1135–1146. doi:10.1056/NEJMra050436.
- Sun C, Huang L, Li Z, et al. Long non-coding RNA MIAT in development and disease: a new player in an old game. J Biomed Sci. 2018;25:23), doi:10.1186/s12929-018-0427-3.
- Sheik MJ, Gaughwin PM, Lim B, et al. Conserved long noncoding RNAs transcriptionally regulated by Oct4 and Nanog modulate pluripotency in mouse embryonic stem cells. RNA. 2010;16:324–337. doi:10.1261/rna.
- Stoyanova E, Trudel M, Felfly H, et al. Vascular endothelial dysfunction in β-thalassemia occurs Despite increased eNOS expression and Preserved vascular Smooth muscle cell Reactivity to NO. PLoS ONE. 2012;7; doi:10.1371/journal.pone.0038089.
- Butthep P, Rummavas S, Wisedpanichkij R, et al. Increased circulating activated endothelial cells, vascular endothelial growth factor, and tumor necrosis factor in thalassemia. Am J Hematol. 2002;70:100–106. doi:10.1002/ajh.10101.
- Aessopos A, Farmakis D, Deftereos S, et al. Thalassemia heart disease: a comparative evaluation of thalassemia major and thalassemia intermedia. Chest. 2005;127:1523–1530. doi:10.1378/chest.127.5.1523.
- Pourfarzad F, von Lindern M, Azarkeivan A, et al. Hydroxyurea responsiveness in β-thalassemic patients is determined by the stress response adaptation of erythroid progenitors and their differentiation propensity. Haematologica. 2013;98:696–704. doi:10.3324/haematol.2012.074492.