
ABSTRACT
Introduction: In sickle cell disease (SCD) patients, among the predictors of survival, HbF levels play a significant role in lowering the morbidity and mortality. Coinheritance of δβ thalassemia and hereditary persistence of fetal hemoglobin (HPFH) may contribute to variable HbF levels in SCD patients, thus influencing their clinicopathological profile. Such cases are sparsely documented in the literature and thus, we screened the presence of δβ thalassemia and HPFH in 126 cases of SCD with high HbF.
Material and methods: A total 126 SCD individuals with raised HbF levels were the study subject. Capillary zone electrophoresis (CZE) was done for the quantitative assessment of hemoglobin variants. HbSC, HbSD, HbAS and HbSE cases were excluded. Asian Indian Gγ(Aγδβ)0-thal, δβ0-thal (Sicilian, 13.4 kb), (Chinese, 100 kb), HPFH-1 (Black, 106 kb), HPFH-2 (Ghanaian, 105 kb), HPFH-3 (Indian, 48.5 kb) were done by GAP-PCR.
Results: Out of 126, 78 cases (62%) were homozygous for SCD. The remaining 48 cases suspected to be heterozygous were furthered screened and 6/48 cases (12.5%) were found to be compound heterozygous. Out of these 6 cases,4(66.66%) had HbS/ δβ- Gγ(Aγδβ)0 and 2(33%) had HbS/HPFH compound heterozygous condition. None of the patients had δβ0-thal (Sicilian, 13.4 kb), (Chinese, 100 kb), HPFH-1 (Black, 106 kb), HPFH-2 (Ghanaian, 105 kb).
Conclusion: This study highlights the importance of understanding the complex patho-physiology of compound heterozygous cases of HbS/HPFH and HbS/δβ thalassemia, as these infrequent conditions lead to change in phenotype and clinical severity of the disease. Insight into more such cases will open the window to better analyze the disease pathogenesis in these rare compound heterozygous conditions, as this will be beneficial to formulate proper management protocol in these patients.
Introduction
Sickle cell disease (SCD) is a hemoglobinopathy, caused by point mutations in the β-hemoglobin gene (HBB), resulting in the substitution of adenine for thymine (GAG > GTG) at codon 6 of the β-globin gene. This results in change in amino acid (valine for glutamic acid) which leads to polymerization of the abnormal sickled hemoglobin i.e. HbS when deoxygenated. As the disease in progresses, there is deformation of the shape of initially reversible deformed red blood cells into the rigid sickle cells which cannot pass through the microcirculation efficiently, resulting anemia, cell adhesion, vaso-occlusion, and ultimately painful sickle cell crisis [Citation1,Citation2]. The sickle cell gene is widespread across in India and it is believed to represent an occurrence of the HbS mutation separate from those in Africa. This is known as the Asian haplotype [Citation3].
Among the predictors of survival in SCD, HbF levels play a very important and significant role in these patients as raised HbF levels may be the cornerstone in lowering down the morbidity and mortality. HbF inhibits HbS polymerization and its abundance in the red blood cells dilutes down the concentration of HbS [Citation4–6]. Different studies have stated that a high level of HbF have protective role or reduce the severity of sickle cell disease. HbF is present at residual levels in healthy adults with over 20-fold variation. Twin studies have shown that 89% of the quantitative variation is heritable but the genetic etiology is complex [Citation7,Citation8]. Measured genotyped analyses have shown the trait to be influenced by several factors including age and sex (which account for 2% of trait variation) and genetic loci linked and unlinked to the b-globin complex. Different types of modulators have been identified which alter clinical severity of SCD, likes HbF levels, alpha thalassemia (α-thal), β-globin gene cluster haplotypes [Citation9,Citation10].
Also, single nucleotide polymorphism (SNP) in HBB gene have been found to be associated in a high level of HbF, usually under conditions of poor erythropoiesis, such as SCD and beta-thalassaemia. The best known of these is a common SNP (C to T) at position −158 of the Gc globin promoter, creating an Xmn1-Gc restriction site. The Xmn1-Gc site (T variant) is common in all population groups and present at a frequency of 0.32–0.35 [Citation11–13].
δβ-thalassemia and hereditary persistence of fetal hemoglobin (HPFH) are also important high Hb F determinants. However, there is sparse data available, especially from India regarding genotype phenotype correlation of δβ-thalassemia and HPFH with sickle cell disease [Citation14,Citation15]. Very few studies from India which have mentioned the compound heterozygous SCD and δβ-thalassemia/ HPFH have been documented till date. In view of this, we conducted this study with aim to investigate the co-existence of Asian Indian inversion-deletion Gγ(Aγδβ)0-thal, δβ0-thal (Sicilian, 13.4 kb), Gγ(Aγδβ)0-thal (Chinese, 100 kb), HPFH-1 (Black, 106 kb), HPFH-2 (Ghanaian, 105 kb), HPFH-3 (Indian, 48.5 kb) in cases of sickle cell disease with a high Hb F concentration ().
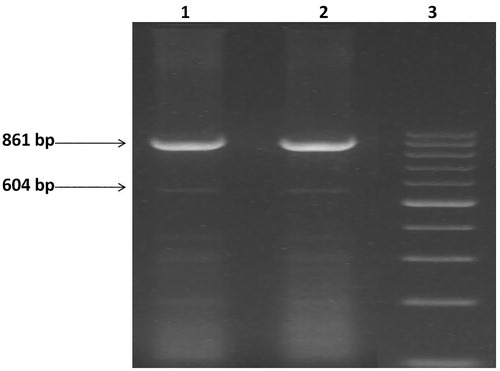
Figure 1. Agarose gel picture showing deletional high persistance of fetal hemoglobin (HPFH-3). Amplification of 861 bp PCR product is for normal whereas 604 bp is deletional HPFH. Lane 1,2, is positive for Indian deletion. Lane 3 is 100 bp marker.
Materials and methods
A total of 126 individuals with raised HbF (>5) levels detected during routine screening at Clinical Hematology OPD, All India Institute of Medical sciences(AIIMS), New Delhi in the last 5 years (year 2013–2017) were the study subjects. A complete history which included the history of jaundice and blood transfusion was taken. Written informed consent was taken from the patients and this study was granted ethical approval from AIIMS, Institute ethical committee. About 10 ml of venous blood was drawn in EDTA vial and complete blood count was measured by automated cell analyzer. Giemsa-stained peripheral blood smears were examined for red cell morphology. Capillary zone electrophoresis (CZE) (SEBIA-PARK TECHNOLOGY, LISSES, FRANCE) was used for the quantitative assessment of hemoglobin variants HbS, HbA, HbF, and HbA2. HbSC, HbSD and HbSE cases were excluded. Genomic DNA was isolated from whole blood using a commercially available DNA isolation kit. Confirmation of the sickle mutation (codon 6, GAG > GTG) was done by amplification refractory mutation system-polymerase chain reaction (ARMS-PCR) [Citation16]. Hemoglobin variant study by CZE was carried out on both parents of all the 126 cases. Screening of six different deletions, these included Asian Indian inversion-deletion δβ-Gγ(Aγδβ)0-thal, δβ0-thal (Sicilian, 13.4 kb), Gγ(Aγδβ)0-thal (Chinese, 100 kb), HPFH-1 (Black, 106 kb), HPFH-2 (Ghanaian, 105 kb), HPFH-3 (Indian, 48.5 kb), were done by GAP-Polymerase Chain Reaction (GAP-PCR). The primer sequences and protocol strategy was followed as described by Craig et al. [Citation17].
Results
Out of 126 cases, 78 cases (62%) were homozygous for Sickle cell disease. The remaining 48 cases (38%) with HbS levels between 60% and 80% on CZE were suspected to be either in heterozygous state or some compound heterozygous condition. On carrying out mutational screening in these 48 cases, we observed 6/48 cases (12.5%) were compound heterozygous condition and the remaining 42(87.5%) cases were HbS trait and were removed from the study. Out of these 6 cases,4 (66.66%) had HbS/ δβ- Gγ(Aγδβ) 0 and 2 (33%) had HbS/HPFH compound heterozygous condition which was confirmed on parental mutational analysis. We observed that for HbS/ δβ- Gγ(Aγδβ) 0 cases, one parent was having sickle cell trait and other was heterozygous for δβ- Gγ(Aγδβ) 0. Similarly, for HbS/ HPFH cases, one parent was having sickle cell trait and other was heterozygous for HPFH.
Clinico-hematological parameters and CZE findings of co-inherited δβ-Gγ(Aγδβ)0 thalassemia and HPFH patients with sickle cell disease is summarized in . Patient 1 and 3 of HbS/δβ-Gγ(Aγδβ) 0 compound heterozygous had more painful crises, frequent blood transfusion and hospitalization than patient 2 and 4. Splenomegaly was present in patient 1and 2. Intracellular distribution of HbF revealed heterocellular distribution in the four compound heterozygotes HbS/δβ-Gγ(Aγδβ)0, while in two cases of HbS/HPFH had pancellular distribution. All the six compound heterozygous had mild microcytic and hypochromic anemia. The highest levels of HbF concentration (29.2 ± 3.8) were observed in HbS/HPFH compound heterozygous ().
Table 1. Hematological indices and clinical history of four cases of βs/ Gγ(Aγδβ)0-thalassemia.
Table 2. Hematological indices and clinical history of two cases of βs/HPFH-thalassemia.
Discussion
SCD is a multigenic trait with diverse clinical and phenotypic variability. Various factors implicated in modification of this disorder include HbF concentration, β-globin gene cluster haplotypes and α-thalassemia which will ultimately influence its clinical severity [Citation9]. Intracellular distribution of Hb F is a strong modifier of sickle cell disease and studies have shown that elevated HbF levels tend to reduce the severity of SCD [Citation10]. Very little data is available regarding the co-expression of HbF modifiers like δβ thalassemia and HPFH with SCD and their influence on the disease severity and phenotype. Higher levels of HbF (Mean 22.3 ± 6.9%; Range 3.9–40.9%) found in Indian SCD patients [Citation18]. The βS gene is linked to an Asian haplotype and the XmnI restriction site, but these factors alone cannot explain the high Hb F concentration [Citation14].
δβ thalassemia and HPFH are the heterogeneous disorders caused by large deletions involving both δ and β globin genes in the β-globin cluster. δβ thalassemia, a form of beta-thalassemia is characterized by decreased or absent synthesis of the δ and β globin chains with a compensatory increase in expression of fetal γ chain synthesis, resulting in augmented production of Hb F in adult life. As homozyzotes have no δ or β genes, they cannot synthesize HbA and HbA2 and have very high HbF, almost approaching 100%. The HPFH include a wide range of conditions characterized by deletional and non-deletional forms of this disorder. Deletional HPFH is more common and among these, HPFH-3 deletion is commonest in India. In HPFH-3, the deletion removes 48.5 kb of a DNA segment, starting from the 5′ end of the ψβ gene to a region 30 kb downstream of the β-globin gene in an L1 repetitive region [Citation14,Citation15,Citation17].
In the present study, 4/6 (66.66%) patients were compound heterozygous for HbS/δβ thalassemia and 2/6 (33%) were compound heterozygous for HbS/HPFH. Among these 4 patients who were compound heterozygous for HbS/δβ thalassemia, HbF levels were 28.5, 32.8, 33.0, and 27.0%, respectively, as compared to mean HbF levels of 22.4% in HbSS patients. These patients had mild microcytic and hypochromic anemia as compared to HbSS patients. HbA2 levels were slightly lower in these patients as compared to HbSS patietns, which is explained by reduced δ chain syntheses in these cases. Mild splenomegaly was also observed among 2/4 patients as compared to absence of splenomegaly in HbSS patients. This could be explained by the fact that in HbSS, there is reduced size of spleen/autosplenectomy in advanced cases Though there was a raised HbF level in these patients as compared to homozygous SCD patients, but we observed more episodes of painful crisis, greater frequency of blood transfusion and increased hospitalization episodes in two patients as compared to HbSS patients. The other two patients had a similar clinical profile when compared with HbSS patients. This can be explained by the possibility of heterocellular distribution of HbF in these patients. Secondly, there could be more complex mechanisms involved in influencing the clinical profile of these patients which further needs to be studied. Our study is in line with Henthorn et al. and Patel et al., and in contrast to Altay et al. [Citation16,Citation19,Citation20]. The variation in these observations warrants the inclusion of more such studies in a larger subset of patients to better understand the patho-physiology of these compound heterozygous patients.
Among the two patients who were compound heterozygous for HbS/HPFH thalassemia, HbF levels were 27% and 29% respectively as compared to mean HbF levels of 22.4% in HbSS patients. We observed lesser episodes of painful crisis, greater frequency of blood transfusion and increased hospitalization episodes in these patients. This can be explained by the possibility of pancellular distribution of HbF in these patients. Our study is in line with Kulozik et al. in Asian population and in contrast to Patel et al. in Indian population [Citation14,Citation20].
In conclusion, this study highlights the importance of understanding the complex patho-physiology of compound heterozygous cases of HbS/HPFH and HbS/δβ thalassemia, as these infrequent conditions lead to change in phenotype and clinical severity of the disease. Insight into more such cases will open the window to better analyze the disease pathogenesis in these rare compound heterozygous conditions, as this will be beneficial to formulate proper management protocol in these patients.
Author’s contribution
HRP designed the study, performed experimental studies and drafted the manuscript. KS compiled the data, and contributed to writing. RR and SP performed analysis and helped in drafting the manuscript. AS and KK reviewed the manuscript and helped in shaping the manuscript. MM, TS and RS provided valuable suggestions and clinical outputs. All authors read and approved the final manuscript.
Disclosure statement
The authors declared no conflict of interest with respect of this article.
Additional information
Funding
References
- Frempong KO, Steinberg MH. Clinical aspects of sickle cell anaemia in adults and children. In: Steinberg MH, Forget BG, Higgs DR, Nagel RL, editors. Disorders of hemoglobin: genetics, pathophysiology, and clinical management. 1st ed. Cambridge: Cambridge University Press; 2001. p. 611–670.
- Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med. 1997;337(11):762–769. doi: 10.1056/NEJM199709113371107
- Kulozik AE, Wainscoat JS, Serjeant GR, et al. Geographical survey of βs-globin gene haplotypes: evidence for an independent Asian origin of the sickle-cell mutation. Am J Hum Genet. 1986;39:239–244.
- Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease: life expectancy and risk factors for early death. N Engl J Med. 1994;330:1639–1644. doi: 10.1056/NEJM199406093302303
- Noguchi C, Schechter AN, Rodgers GP. Sickle cell disease pathophysiology. In: DR Higgs, DJ Weatherall, editors. Bailliere’s clinical haematology: the haemoglobinopathies. Vol. 6. London: Bailliere Tindall; 1993. p. 57–91.
- Thein SL. Beta thalassaemia. In: GP Rodgers, editor. Bailliere’s clinical haematology, sickle cell disease and thalassaemia, Vol. 11:1. London: Bailliere Tindall; 1998. p. 91–126.
- Garner C, Tatu T, Reittie JE, et al. Genetic influences on F cells and other hematologic variables: a twin heritability study. Blood. 2000;95:342–346. doi: 10.1182/blood.V95.1.342
- Garner C, Tatu T, Game L, et al. A candidate gene study of F cell levels in sibling pairs using a joint linkage and association analysis. GeneScreen. 2000;1:9–14. doi: 10.1046/j.1466-9218.2000.00001.x
- Steinberg MH. Predicting clinical severity in sickle cell anaemia. Br J Haematol. 2005;129(4):465–481. doi: 10.1111/j.1365-2141.2005.05411.x
- Gilman JG, Huisman THJ. DNA sequence variation associated with elevated fetal Gc globin production. Blood. 1985;66:783–787. doi: 10.1182/blood.V66.4.783.783
- Labie D, Pagnier J, Lapoumeroulie C, et al. Common haplotype dependency of high G gamma-globin gene expression and high HbF levels in beta-thalassemia and sickle cell anemia patients. Proc Natl Acad Sci USA. 1985;82:2111–2114. doi: 10.1073/pnas.82.7.2111
- Thein SL, Wainscoat JS, Sampietro M, et al. Association of thalassaemia intermedia with a beta-globin gene haplotype. Br J Haematol. 1987;65:367–373. doi: 10.1111/j.1365-2141.1987.tb06870.x
- Weatherall DJ, Clegg JB, editors. Hereditary persistence of fetal hemoglobin. The thalassaemia syndromes. 4th ed. Oxford: Blackwell Science; 2001. p. 450–483.
- Kulozik AE, Kar BC, Satapathy RK, et al. Fetal hemoglobin levels and βS globin haplotypes in an Indian population with sickle cell disease. Blood. 1987;69(6):1742–1746. doi: 10.1182/blood.V69.6.1742.1742
- Wu Y, Ugozzoli L, Pal BK, et al. Allele-specific enzymatic amplification of β-globin genomic DNA for diagnosis of sickle cell anemia. Proc Natl Acad Sci. 1989;86(8):2757–2760. doi: 10.1073/pnas.86.8.2757
- Henthorn PS, Smithies O, Nakatsuji T, et al. (Aγδβ)0-thalassaemia in blacks is due to a deletion of 34 kbp of DNA. Br J Haematol. 1985;59(2):343–356. doi: 10.1111/j.1365-2141.1985.tb02999.x
- Craig JE, Barnetson RA, Prior J, et al. Rapid detection of deletions causing δβ thalassemia and hereditary persistence of fetal hemoglobin by enzymatic amplification. Blood. 1994;83(6):1673–1682. doi: 10.1182/blood.V83.6.1673.1673
- Mashon RS, Dash PM, Khalko J, et al. Higher fetal hemoglobin concentration in patients with sickle cell disease in eastern India reduces frequency of painful crisis. Eur J Haematol. 2009;83(4):383–384. doi: 10.1111/j.1600-0609.2009.01290.x
- Altay Ç, Schroeder A, Huisman THJ. The Gγ-δβ-thalassemia and Gγ-β0-HPFH conditions in combination with β-thalassemia and Hb S. Am J Hematol. 1977;3(1):1–14. doi: 10.1002/ajh.2830030101
- Patel DK, Patel M, Mashon RS, et al. Clinical and molecular characterization of β(S) and (G)γ((A)γδβ)⁰-thalassemia in eastern India. Hemoglobin. 2010;34(6):604–609. doi: 10.3109/03630269.2010.526890