
ABSTRACT
Objective: To study the gene mutation of human coagulation factor XII (FXII) in a Chinese family with FXII deficiency and it will help us to understand the pathogenesis of this type of disease.
Clinical presentation: The proband was a 50-year-old male who had a fracture of the right humerus. The routine presurgical coagulation test showed a significant prolonged activated partial thromboplastin time (APTT) at 59.1s (reference range, 29.0–43.0s).
Techniques: FXII activity (FXII:C) and FXII antigen (FXII:Ag) were detected by the one-stage clotting method and ELISA, respectively. To identify mutations, the FXII whole exon and flanking sequences were carried out. Suspected mutations were confirmed by reverse sequencing. The conservatism and possible impact of the amino acid substitution were analyzed by ClustalX-2.1-win and four online bioinformatics tools.
Results: Phenotypic analysis revealed the FXII:C and FXII:Ag of the proband were 4% and 5%, respectively (normal range, 72–113%). Gene sequencing detected compound heterozygous mutations c.1561G > A (Glu502Lys) and c.1637T > C (Met527Thr) in exon 13. Bioinformatics and model analysis indicated that mutations probably had disrupted the function and structure of the FXII protein.
Conclusion: We detected two missense mutations Glu502Lys and Met527Thr in the catalytic domain of the proband, of which Met527Thr was first reported in the world. Our findings suggest that the double mutations in the FXII gene were the causing reasons for the decreased FXII:C and FXII:Ag. These results not only enriched the F12 mutation database in this condition, but also helped to identify the genetic defects of FXII in China.
Introduction
Human coagulation factor XII (FXII) is a serine protease precursor mainly synthesized by the liver. The mature protein consisting of 596 amino acid residues, has a concentration of 30–35 ug mL−1 in circulating plasma and a half-life of 50–70 h [Citation1]. The gene encoding FXII is 12 kb in size, located on the chromosome 5q33-qter and consists of 14 exons. FXII plays an important role in initiating the endogenous pathway of blood coagulation. When it contacts the negatively charged surface in vitro, the attached precursor is cleaved by kallikrein at Arg353-Val354, which in turn activates more FXII protein [Citation2,Citation3]. Activated FXII (FXIIa) not only initiates the endogenous coagulation pathway, but also participates in activation of the fibrinolysis pathway, complement activation, and inflammatory response [Citation4–6].
Congenital FXII deficiency is a rare autosomal recessive disorder. Although it leads to a significant prolongation of activated partial thromboplastin time (APTT) in vitro, patients do not show significant bleeding tendency [Citation7], so most of them were accidentally discovered by health check or preoperative coagulation screening tests [Citation8,Citation9]. Several clinical investigations have shown that FXII deficiency was a risk factor for thrombosis [Citation10–12]. However, animal studies have found that despite normal hemostasis, the risk of thrombosis (such as arterial thrombosis) in FXII - / - mice was reduced [Citation13,Citation14]. FXII may be an essential element of thrombosis in vivo [Citation14]. Many reported cases of FXII deficiency have no thrombosis, which is consistent with animal studies[Citation4,Citation10,Citation15,Citation16]. These findings have generated new interest in FXII because it affects thrombosis without affecting hemostasis, so FXII may be the perfect target for the development of new antithrombotic drugs [Citation17].
Four bases upstream of the initiation ATG codon, 46 C to T in exon 1 of the 5′-untranslated region, is a common genetic polymorphism of FXII, which can be divided into 46CC, 46CT, 46TT genotypes. Studies have shown that this polymorphism associated with low translational efficiency and decreased plasma FXII levels [Citation18].
FXII deficiency can be divided into two categories based on FXII activity and antigen levels in plasma. Type I disorders (CRM-negative), decreased activity with the feature of low plasma FXII antigen levels. Most of the reported cases fall into this category. And type II disorders (CRM-positive), which is characterized by decreased activity but borderline-normal or normal antigen.
Here we report an FXII-deficient patient, and we studied the phenotype and genotype of the proband and his families. Two amino acid substitutions, Glu502Lys and Met527Thr, were found in the catalytic domain, of which Met527Thr was the first report in the world.
Patients and methods
Patients
Subjects under study were the proband, his father, mother, spouse, and daughter, a total of five members of three generations (). The proband was a 50-year-old Chinese male who admitted to our hospital due to traumatic fracture of his right humerus. He had no history of abnormal bleeding or thrombosis. The routine preoperative examination revealed a prolongation of APTT (59.1s, reference range: 29.0–43.0s) and further factor assays showed decreased FXII activity (FXII:C) and FXII antigen (FXII:Ag) at 4% and 5% (reference range, 72–130%), respectively, the proband was diagnosed with ‘FXII deficiency, fracture of right humerus.’ The fracture site was treated with open reduction and internal fixation, the operation proceeded smoothly without bleeding or thrombosis.
Figure 1. The family tree of the inherited coagulation FXII deficiency.
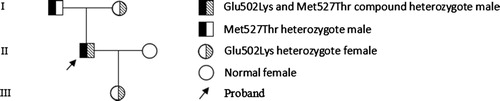
We took the peripheral blood of other four family members in order to confirm the cause of FXII deficiency. Coagulation tests showed that the FXII:C and FXII:Ag of his father, mother and daughter were decreased to approximately 30% and his spouse with normal FXII:C and FXII:Ag. We also tested the APTT, prothrombin time (PT), thrombin time (TT), and fibrinogen (FIB) of the four family members and found no abnormal results (the data of PT, TT and FIB were not shown).
Methods
Coagulation index detection
Our study was approved by the Ethics Committee of the First Affiliated Hospital of Wenzhou Medical University (China). With the informed consent, blood samples were collected from the patient and his family members. APTT, PT, TT, FIB and FXII: C were tested by a clotting assay on the STAGO-STAR analyzer (DIAGNOSTICA STAGO, Asnière sur Seine, France) using matched commercially available kits. And the FXII:Ag was detected by enzyme linked immunosorbent assay (ELISA) kit (Changfeng, Wenzhou, China), in which polyclonal goat anti-human FXII IgG was used as coating antibody and monoclonal peroxidase-conjugated goat anti-human FXII IgG was used as detecting antibody. All operations were performed in accordance with the manufacturer’s instructions.
DNA extraction and PCR amplification
DNA specimens for genotypic analysis were extracted from anticoagulant blood samples according to the manufacturer’s protocol (Tiangen, Beijing, China). We designed seven pairs of primers containing all exons and flanking regions of the F12 gene and the primer sequences were listed in . PCR was amplified under standard conditions on Applied Biosysestem Thermal Cycler 2720 (ABI, Foster City, California, U.S.A.). And the purified PCR products were directly sequenced at Sunsoon BIO-Technology Corporation (Shanghai, China). 100 individuals were selected to exclude the polymorphism.
Table 1. Primer sequences and amplified fragment length of factor XII gene.
Conservation analysis and Bioinformatics prediction
We used the multi-sequence alignment software ClustalX-2.1-win (Science Foundation Ireland, Dublin, Ireland) to analyze the conserved properties of Gln502 and Met527 in homo sapiens and seven homologous species: Mus musculus, Rattus norvegicus, Pan troglodytes, Macaca mulatta, Canis lupus familiaris, Bos Taurus, and Xenopus tropicalis (HomoloGene, http://www.ncbi.nlm.nih.gov/homologene). Four online bioinformatics softwares (Mutation Taster, PolyPhen-2, PROVEAN and SIFT) were used to predict whether amino acid substitution would affect the function and structure of FXII protein. Molecular structure of the mutant FXII was analyzed by Swiss-Pdb Viewer version 4.0.1. (Swiss Institute of Bioinformatics, Lausanne, Switzerland) and PIC programs (Protein Interactions Calculator, http://pic.mbu.iisc.ernet.in) based on the three-dimensional (3D) structure in the Protein Data Bank (PDB, http://www.rcsb.org/pdb/home/home.do, PDB ID: 4XDE).
Results
Coagulation tests
The phenotype and genotype of all the family members are shown in , and proband is shown in bold font. The proband had no history of abnormal bleeding or thrombosis. Except the APTT was raised at 59.1s (normal range: 29.0–43.0s), and FIB was slightly raised at 4.44 g/L (normal range: 2–4 g/L), other indexes such as the platelet levels, PT, TT were within normal ranges. Factor assays showed that the FXII:C levels of the proband, his father, mother and daughter were reduced to 4%, 32%, 36% and 35%, respectively, parallel decrease in FXII:Ag was the same as FXII:C, remained at 5%, 34%, 37% and 36%, respectively. FXII:C and FXII: Ag of the proband’s spouse were within normal range.
Table 2. Phenotypic and genetic analysis of the pedigree of family members.
DNA sequence analysis
Sequence analysis of F12 gene revealed the proband took compound heterozygous mutations in exon 13. One was a c.1561G > A point mutation resulting in an amino acid substitution of glutamate (GAG) to lysine (AAG). Another was a c.1637T > C point mutation resulting in a methionine (ATG) to threonine (ACG) substitution. His father, mother and daughter were carried a heterozygous c.1637T > C, c.1561G > A and c.1561G > A point mutation, respectively. Coagulation index detection consequences were in line with genetic results. These two mutations were absent in 100 normal controls. As for the 46CT polymorphism, except the proband and his father were heterozygotes of 46CT, all other three family members were 46CC wild types.
Bioinformatics analysis
Homologous sequence alignment results showed that both Glu502 and Met527 were highly conserved within Homo sapiens, Pan troglodytes, Macaca mulatta, Canis lupus familiaris, Bos taurus, Mus musculus, Rattus norvegicus and Xenopus tropicalis ().
Figure 2. Conservative analysis diagrams. The targeted amino acid was indicated with an arrow.

The predicted results of the Glu502Lys were ‘disease causing’, ‘benign’, ‘deleterious’ and ‘affect protein function’, corresponding to ‘Mutation Taster’, ‘PolyPhen-2’, ‘PROVEAN’, and ‘SIFT’; and the consequence of Met527Thr were ‘disease causing’, ‘probably damaging’, ‘deleterious’ and ‘affect protein function’ according to the four softwares ().
Table 3. The results of the analysis of bioinformatics online softwares.
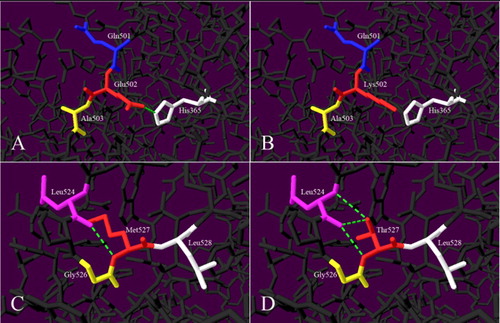
Analysis of the 3D protein model indicated that substitution of Glu502 with Lys502 resulted in the disappearance of hydrogen bonds between Glu502-His365. Replacement of Met527 with Thr527 resulted in the addition of two hydrogen bonds between Thr527-Leu527 ().
Figure 3. Model analysis diagrams. (A–B) Glu502Lys, (C–D) Met527Thr before and after mutations in turn. The green dotted lines indicate hydrogen bond.
Discussion
According to the results of the family study, the proband’s FXII:C and FXII:Ag were reduced to approximately 4%, and he took compound heterozygous mutations Glu502Lys and Met527Thr in exon 13 of F12. While his father, mother and daughter’s FXII: C and FXII: Ag were reduced to approximately 30% and they carried heterozygous mutations of Met527Thr, Glu502Lys and Glu502Lys, respectively. His spouse’s FXII: C and FXII: Ag were normal, and she was the wild type of Glu502 and Met527. In addition, expect the proband and his father were heterozygotes 46CT, the other three members all took 46CC genotype, which indicated that the 46CT polymorphism has no significant effect on the reduction of FXII:C in this family. There are no other factors contribute to the activity and antigen reduction of FXII, and the reduced degree of FXII:C and FXII:Ag in the compound heterozygous FXII deficiency is consistent with many previous reports [Citation15,Citation16]. Therefore, we initially believe that the compound heterozygous mutations Glu502Lys and Met527Thr were responsible for the reduction of FXII:C and FXII:Ag.
Hereditary FXII deficiency is a rare autosomal recessive coagulation disorder, and individuals with severe FXII deficiency have markedly prolonged APTT in vitro but no bleeding tendency in vivo [Citation19]. Based on previous research results [Citation12], we know that the compound heterozygotes have a lower degree of activity and antigen levels compared to the single heterozygote. In addition, almost all compound heterozygotes showed the same phenotypic characteristics as homozygous mutations, namely very low FXII activity and antigen.
The FXII: C and FXII: Ag of proband were significantly synchronous reduced is type I deficiency (CRM-negative) (), suggesting the synthesis, secretion, and function of the mutant protein were impaired [Citation20]. In combination with , we could see that the compound heterozygous mutations (Glu502Lys and Met527Thr) of proband were inherited from his father and mother, respectively, and the mode of its inheritance is consistent with the common autosomal recessive inheritance pattern of patients with hereditary FXII deficiency. This is the first report of Met527Thr, and Glu502Lys has been reported [Citation15,Citation16].
To further demonstrate the adverse effects of Glu502Lys and Met527Thr, we analyzed their conservatism and possible effects on the protein. According to the results of conservation analysis (), we know that the Glu502Lys and Met527Thr were highly conserved among all homologous species, which indicated that they might play vital roles in the normal function of FXII. The result of online bioinformatics tools () indicates that the Glu502 and Met527 were deleterious mutations and could affect the function and structure of FXII, which is consistent with their conservative property.
Both Glu502 and Met527 were located in the catalytic domain of the FXII protein light chain (catalytic triad: His393-Asp442-Ser544), of which Met527Thr near the active Ser544. Intensive studies have shown that the catalytic domain of FXII protein contains from 373 to 614 residues, which is a key part of the FXII enzyme function. To analyze the influence of mutations on the spatial structure of the protein, we performed the model analysis (). For Glu502Lys, Glu502 had a hydrogen bond with His365. Once replaced by Lys502, the hydrogen bond formed with His365 disappeared. As to the Met527Thr, a hydrogen bond existed between Met527 and Leu524. When Met527 was replaced by Thr527, two additional hydrogen bonds with Leu524 were added. Since hydrogen bonding has a significant effect on maintaining the normal spatial conformation and stability of the protein, these changes may affect the subtle spatial structure of the catalytic region and ultimately affect the activation and activity of FXII.
In vitro expression study of Glu502Lys indicated that the mutant protein was extensively degraded in the pre-Golgi region of the cell, resulting in a low expression level of the mutein [Citation16]. Functional studies of the mutant FXII protein (Gly531Glu) indicated that this mutation results in partial defects in the cleavage activity of the prekallikrein [Citation3]. Since Met527 is located near Gly531, we hypothesized that the mutant protein caused by Met527Thr has the same functional defect as Gly531Glu.
Up to now, 54 mutations of FXII have been registered in The Human Gene Mutation Database (HGMD, http://www.hgmd.cf.ac.uk/ac/gene.php?gene = FXII), including missense/nonsense, splicing, regulatory, small insertions, small deletions, and gross deletions mutations. Fifteen mutations have been reported in the FXII catalytic region, and none of them have a tendency to bleed. The compound heterozygous mutations (Glu502Lys and Met527Thr) reported in this paper enriched the database.
In summary, we found two missense mutations (Glu502Lys and Met527Thr) in a patient with FXII deficiency, which may alter the spatial structure of the catalytic domain, resulting in decreased activity and antigen of FXII. We preliminarily analyzed their molecular pathogenesis, which will help to further research on FXII.
Acknowledgements
We are grateful to the patient and his family members for cooperation. All authors stated that they had no interests which might be perceived as posing a conflict or bias. All authors have made substantial contributions to the conception and design of the study, acquisition of data, or analysis and interpretation of the data, drafting the article or revising it critically for important intellectual content, and have given their final approval for the version to be submitted.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Bjorkqvist J, Nickel KF, Stavrou E, et al. In vivo activation and functions of the protease factor XII. Thromb Haemost. 2014;112(5):868–875. doi: 10.1160/th14-04-0311
- Dunn JT, Silverberg M, Kaplan AP. The cleavage and formation of activated human Hageman factor by autodigestion and by kallikrein. J Biol Chem. 1982;257(4):1779–1784.
- Iijima K1, Arakawa Y, Sugahara Y, et al. Factor XII Osaka: abnormal factor XII with partially defective prekallikrein cleavage activity. Thromb Haemost. 2011;105(3):473–478. doi: 10.1160/TH10-02-0123
- Cheng X, Yang L, Huang G, et al. Genetic analysis of a hereditary factor XII deficiency pedigree of a consanguineous marriage due to a homozygous FXII gene mutation: Gly341Arg. Hematology. 2017;22(5):310–315. doi: 10.1080/10245332.2016.1265210
- Schmaier AH. The elusive physiologic role of factor XII. J Clin Invest. 2008;118:3006–3009.
- Müller F1, Renné T. Novel roles for factor XII-driven plasma contact activation system. Curr Opin Hematol. 2008;15:516–521. doi: 10.1097/MOH.0b013e328309ec85
- Lämmle B, Wuillemin WA, Huber I, et al. Thromboembolism and bleeding tendency in congenital factor XII deficiency - a study on 74 subjects from 14 Swiss families. Thromb Haemost. 1991;65(2):117–121. doi: 10.1055/s-0038-1647467
- Saito H. Contact factors in health and disease. Semin Thromb Hemost. 1987;13:36–49. doi: 10.1055/s-2007-1003474
- Singhamatr P, Kanjanapongkul S, Rojnuckarin P. Molecular analysis of factor XII gene in Thai patients with factor XII deficiency. Blood Coagul Fibrinolysis. 2013;24(6):599–604. doi: 10.1097/MBC.0b013e32835fde9d
- Kwon MJ, Kim HJ, Lee KO, et al. Molecular genetic analysis of Korean patients with coagulation factor XII deficiency. Blood Coagul Fibrinolysis. 2010;21(4):308–312. doi: 10.1097/MBC.0b013e32833449df
- Kanaji T, Kanaji S, Osaki K, et al. Identification and characterization of two novel mutations (Q421 K and R123P) in congenital factor XII deficiency. Thromb Haemost. 2001;86(6):1409–1415. doi: 10.1055/s-0037-1616743
- Lombardi AM, Bortoletto E, Scarparo P, et al. Genetic study in patients with factor XII deficiency: a report of three new mutations exon 13 (Q501STOP), exon 14 (P547L) and -13C > T promoter region in three compound heterozygotes. Blood Coagul Fibrinolysis. 2008;19(7):639–643. doi: 10.1097/MBC.0b013e32830d8629
- Pauer HU1, Renné T, Hemmerlein B, et al. Targeted deletion of murine coagulation factor XII gene-a model for contact phase activation in vivo. Thromb Haemost. 2004;92(3):503–508. doi: 10.1160/TH04-04-0250
- Renné T, Pozgajová M, Grüner S, et al. Defective thrombus formation in mice lacking coagulation factor XII. J Exp Med. 2005;202(2):271–281. doi: 10.1084/jem.20050664
- Yang L, Wang Y, Zhou J, et al. Identification of genetic defects Underlying FXII deficiency in four Unrelated Chinese patients. Acta Haematol. 2016;135(4):238–240. doi: 10.1159/000444209
- Jin P, Jiang W, Yan H, et al. Novel mutations in congenital factor XII deficiency. Front Biosci (Landmark Ed). 2016;21:419–429. doi: 10.2741/4422
- Li J, Guan X, Liu O, et al. Changes in coagulation factor XII and its function during aortic arch surgery for acute aortic dissection-a prospective observational study. J Thorac Dis. 2018;10(7):4006–4016. doi: 10.21037/jtd.2018.06.127
- Oguchi S, Ishii K, Moriki T, et al. Factor XII Shizuoka, a novel mutation (Ala392Thr) identified and characterized in a patient with congenital coagulation factor XII deficiency. Thromb Res. 2005;115(3):191–197. doi: 10.1016/j.thromres.2004.08.027
- Schloesser M, Zeerleder S, Lutze G, et al. Mutations in the human factor XII gene. Blood. 1997;90(10):3967–3977.
- Suzuki K, Murai K, Suwabe A, et al. Factor XII Ofunato: Lys346Asn mutation associated with blood coagulation factor XII deficiency causes impaired secretion through a proteasome-mediated degradation. Thromb Res. 2010;125:438–443. doi: 10.1016/j.thromres.2009.12.004