
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
Background: The clinical consequences and significance of many unstable hemoglobins interacting with other hemoglobinopathies remain unrecognized. Here we first explore molecular and hematological characterizations of previously undescribed compound heterozygosity states for unstable hemoglobin Rush (Hb Rush, Beta 101 Glu > Gln, HBB:c.304G > C) with Hb E and different forms of thalassemia.
Methods: Hematological assays, globin gene mutation assays and β-globin gene cluster haplotype were conducted in 11 patients from 8 unrelated Chinese ethnic families with unexplained hemoglobin separation fraction in hemoglobin gel electrophoresis.
Results: Hb Rush in various combinations with Hb E, β0-thalassemias and α+-thalassemia were identified. Hb Rush simple heterozygote was generally associated with mild hemolytic anemia, and the compound heterozygotes of Hb Rush and the other β-globin variants led to thalassemia intermedia phenotypes with moderate anemia. Hemoglobin electrophoreses showed that the co-presence of Hb Rush with either Hb E or β0-thalassemias increased proportion of Hb Rush due to relative decrease of other globin chain synthesis. Beta-globin gene cluster haplotype analysis suggested a common origin of the Hb Rush variant in the Chinese families of different ethnic ancestry.
Conclusions: Unstable Hb Rush interacting with β-thalassemia result in thalassemia intermedia phenotypes, which demonstrated the clinical significance of Hb Rush and new insights into complex mechanism of clinical heterogeneity of thalassemia.
1. Introduction
Unstable hemoglobins include a large group of abnormal hemoglobins resulting from the synthesis of structurally abnormal globin chains with varying degrees of hemoglobin molecular instability. Approximately 150 unstable hemoglobin variants have been reported to date (http://globin.bx.psu.edu/hbvar). Most patients with unstable hemoglobins present with only mild hemolytic anemia or are asymptomatic, thus causing these individuals to be frequently undiagnosed and untreated in clinical practice. However, some unstable hemoglobins in association with β-thalassemia can result in the presentation of thalassemia intermedia or even thalassemia major [Citation1,Citation2], thus demonstrating that unstable hemoglobins and thalassemia can modify each other’s clinical phenotypes and cause severe clinical manifestation. The interaction of unstable hemoglobin and thalassemia represents a category of genotypes encountered in thalassemia intermedia [Citation3]. However, the clinical features and mechanisms of the interactions between unstable hemoglobins and thalassemia are exceptional and still poorly understood.
Unstable hemoglobin (Hb) Rush [Beta 101(G3) Glu > Gln, HBB:c.304G > C] is one of the early identified hemoglobin variants has been described to be responsible for mild hemolytic anemia in an African-American family and a Brazilian family of Italian descent [Citation4,Citation5]. As only a few Hb Rush simple heterozygotes have been reported in the two isolated families, many characteristics about its clinical features still remain unclear. Furthermore, the functional consequences of Hb Rush interacting with other globin gene mutations have not been recognized. Here, we investigated the hematological and molecular characterizations of both Hb Rush simple heterozygote and compound heterozygotes of Hb Rush variant with thalassemia mutations, in ethnic families from southwestern China where hemoglobinopathies are very common [Citation6,Citation7]. We provide the first evidence of Hb Rush in association with Hb E and β0-thalassemia mutations leading to thalassemia intermedia phenotypes.
2. Materials and methods
2.1. Participants
Eleven patients from 8 unrelated ethnic families in Yunnan province, Southwestern China, were enrolled in this study (). The probands were discovered initially via a retrospective review of hematological test records with unexplained hemoglobin migration fractions in hemoglobin gel electrophoresis, which were subsequently identified through molecular genetic analyses. Family members were investigated whenever possible. Some patients’ data and blood were retrieved from the local clinical database, therefore information on every clinical feature and laboratory test was not available for all participants. Informed consent according to the Declaration of Helsinki was obtained from all participants. Ethical approval for the study protocol was obtained from the Ethics Committee Board of the Institute of Medical Biology, Chinese Academy of Medical Sciences.
Table 1. Hematological and molecular genetic data of the study subjects.
2.2. Hematological and hemoglobin analysis
Hematological parameters of anticoagulated peripheral blood were measured with an automated hematology analyzer (Sysmex, Kobe, Japan). Hemoglobin fraction separations were performed with agarose gel electrophoresis in an alkaline (pH 8.0) medium in an Interlab G26 clinical electrophoresis instrument (Interlab SRL, Rome, Italy), and a Capillarys 2 Flex-Piercing system (Sebia Benelux SCS, Vilvoorde, Belgium) in a pH 9.4 alkaline buffer.
2.3. Molecular genetic analyses
Genomic DNA was extracted from peripheral blood. Deletional and non-deletional α-thalassemia mutations were examined using multiplex Gap-PCR and amplification refractory mutation system (ARMS), respectively, as described previously [Citation8,Citation9]. Beta-globin gene (HBB) variants were identified via amplification of 1973bp of an HBB gene fragment including all exons and partial untranslation regions (UTR) by using PCR followed by Sanger DNA sequencing (Supplementary table 1). By following the guidelines for ARMS technique development [Citation10], we developed a tetra-primer ARMS-PCR protocol for the rapid detection of the G > C single allelic variant of Hb Rush (Supplementary table 2 and supplementary figure 1). In brief, PCR was performed in a single tube containing total four primers (tetra-primer): two allele-specific primers (inner primers) respectively against wild type allele G and mutant allele C at HBB:c.304 position, and two outer primers asymmetrically paired with the inner primers. Allele specific PCR fragments with 305 bp for G allele and 108 bp for C allele were generated by optimized PCR conditions and then separated by agarose gel electrophoresis. To explore the origin of Hb Rush variant in the different ethnic families, β-globin gene cluster haplotypes were defined on the basis of seven polymorphic restriction sites: HincII in 5′ to ε-globin, HindIII in G γ and Aγ-globin gene, HincII in 5′ to Ψβ and its 3′ region, AvaII in IVS2 of β and Hinf I in 3′ to β globin gene, by using PCR-RFLP (Restriction fragment length polymorphism) protocols, as previously described [Citation11,Citation12].
3. Results
3.1. The coinheritance of Hb Rush with Hb E and thalassemia were identified in the Chinese ethnic families
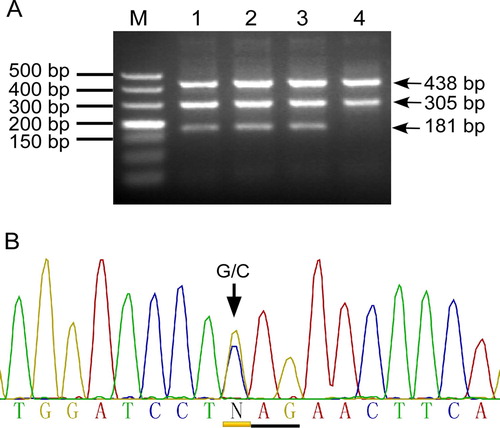
Hb Rush mutation in the participants was identified by using the tetra-ARMS method we have developed (A). DNA sequencing confirmed the Hb Rush GAG > CAG (Glu > Gln) mutation at HBB gene codon 101 (HBB: c. 304G > C) and the heterozygous state of the mutation in all of the detected cases (B). No homozygous Hb Rush variant was found. We also found that Hb E (beta 26, Glu < Lys, HBB:c.79G > A), β0-thalassemia mutations CD17 (Beta 17, Lys > Stop, HBB:c.52A > T) and CD72 (Beta 72 + A, Phe Ser > Stop, HBB:c.216_217insA), and a deletional α+-thalassemia mutation −α3.7 in association with the Hb Rush variant in various combinations (). We determined 5 Hb Rush single heterozygotes, 4 compound double heterozygotes (α−3.7 heterozygote/βRush, α−3.7 homozygote/βRush, βE/βRush, βCD72/βRush) and 2 compound triple heterozygotes of Hb Rush and the other hemoglobinopathies variants (α−3.7/βE/βRush, α−3.7/βCD17/βRush) in the 11 patients from 8 unrelated Chinese families of Han, Thai and Jingpo descents. We detected the Hb Rush variant in different Chinese families from Southwest China and found the co-inheritance of Hb Rush with Hb E, α+- and β0-thalassemia mutations. These results showed that Hb Rush is distributed in different Chinese ethnic populations in Southwest China and interacts with other hemoglobin disorders.
Figure 1. Identification of the Hb Rush mutation using tetra-primer ARMS. A: Representative agarose gel electrophoresis pattern of tetra-primer ARMS for detecting the Hb Rush G > C mutation. M: DNA ladder; Lanes 1–3, heterozygote of Hb Rush mutation, which produces a 181 bp fragment specific for the mutant allele C, a 305-bp fragment for wild-type allele G, and a 438-bp fragment for internal control. Lane 4, normal control. B: Sanger DNA sequencing confirmed the Hb Rush G/C heterozygous state.
3.2. Hb Rush variant resulted in thalassemia intermedia phenotypes in association with Hb E and β0-thalassemia
We analyzed the hematological characterizations of the simple and compound heterozygotes of Hb Rush on the basis of the major hematological findings listed in . We found that Hb Rush simple heterozygote (βA/βRush) generally presented with mild hemolytic anemia with hemoglobin concentrations of 85–109 g/L. However, heterozygote can be asymptomatic (subject P6) as well, indicating that Hb Rush can be a silent mutation in affected participants. An increased reticulocyte percentage (4.4%) was observed in one tested heterozygote (P5), thus indicating a hematopoiesis response to stimulation by mild anemia.
We found that compound heterozygosity states for Hb Rush coinheritance with Hb E (Patients P1 and P7) or β0-thalassemia (P2 and P8) exhibited moderate microcytic hypochromic anemia with decreased HBG (60–93 g/L), MCV (68.5–78.2 fL) and MCH (19.9–24.8 pg). An increased reticulocyte percentage (6.3%) was observed in one compound heterozygote (P7). The anemia of Hb Rush co-inherited with β0-thalassemia tended to be severer than that of Hb Rush co-inherited with Hb E, but none of these patients required transfusion or splenectomization. These results showed that Hb Rush concurrent with Hb E or β0-thalassemia led to β-thalassemia intermedia (TI) syndromes with moderate anemia. We found that all Hb Rush carriers exhibited increased RDW-CV (Red blood cell distribution width, as measured by coefficient of variance), a typical feature of thalassemia presented on red blood cells. The compound heterozygotes of Hb Rush and β0-thalassemia exhibited higher RDW-CV values (24.6–26.0%) than the other combinations (16.0–22%), indicating severer anemia resulted by the compound mutations. These results demonstrated the Hb Rush variant acts in synergy with β0-thalassemia on exacerbating the clinical phenotypes of β-thalassemia. We also found that Hb Rush co-inherited with a homozygote of α+-thalassemia mutation α−3.7 (Patient M2) was associated with α-thalassemia minor phenotypes with mild microcytic hypochromic anemia, and co-inheritance with the α−3.7 heterozygote (Patient M1) was associated with very mild normocytic anemia, showing that the α+-thalassemia mutation does not cooperate with the β-globin variant Hb Rush.
Furthermore, significant variability in hemoglobin concentration (HGB) was observed in Hb Rush carriers subjected to more than one test, and this variability was generally associated with infections and fever. This result indicated the instability nature of the unstable hemoglobin variant, in which environmental factors contribute to the clinical heterogeneity of Hb Rush carriers. We also observed increased reticulocyte indexes of Ret% (reticulocyte percentage), IRF% (immature reticulocyte fraction percentage) and HFR% (high fluorescent reticulocyte percentage) in several tested patients (data not shown), thus indicating that the increased erythropoietic activity in response to the mild hemolytic anemia of Hb Rush single heterozygote and the moderate anemia of thalassemia intermedia resulted from the compound heterozygote states of Hb Rush/Hb E and Hb Rush/β0-thalassemia.
3.3. Compound heterozygotes Hb Rush/Hb E and Hb Rush/β0-thalassemia exhibited decreased Hb A2 and relative increased Hb Rush fractions in hemoglobin separation patterns
To characterize the hemoglobin electrophoresis profiles of the Hb Rush simple heterozygote and the compound heterozygotes, blood samples were analyzed by using agarose gel electrophoresis and capillary electrophoresis whenever possible ( and ). Agarose gel electrophoresis in alkaline (pH 8.0) media revealed that the Hb Rush simple heterozygote (A/Rush) had three distinct bands (A): a more cathodic band of Hb Rush dimer, an intermediate band of hybrid tetramer that closely migrated in the Hb F region, and a more anodic band of HbA (
). Therefore, the Hb Rush fractions were not distinguishable from Hb F in agarose gel electro
phoresis. We also noted that as Hb Rush coinherited with Hb E or β0-thalassemia mutations CD72 and CD17, the Hb A and
tetramer fractions decreased significantly or completely disappeared, while the Hb Rush fraction (
) relatively increased. These results indicated that both β0-thalassemia and Hb E interacted with Hb Rush increased the formation of Hb Rush bearing a beta-globin chain, thereby consequently decreasing normal beta chain production. For the first time, at the Hb molecular level, the cooperation between Hb Rush variant and the βE or β0 alleles in the compound heterozygosity states were presented.
Figure 2. Representative hemoglobin separation profiles of Hb Rush carriers. A: Agarose gel electrophoresis shows hemoglobin separation patterns of normal β alleles (A/A), heterozygote of Hb E (A/E), compound heterozygotes of Hb Rush with β0-thalassemia (Rush/CD17 and Rush/CD72) and Hb E (Rush/E), and simple heterozygote of Hb Rush (Rush / A). Hb Rush dimer and tetramer fractions migrate into the Hb F region. B: Capillary electrophoretic pattern of a simple heterozygote of Hb Rush. Hb Rush migrates into the Z8 zone. C: Capillary electrophoretic pattern of a compound heterozygote of Hb Rush and Hb E showing a significant proportion of Hb Rush and absent Hb A distinguishing from slightly increased Hb F. Hemoglobin fractions and migration zones were not automatically determined, owing to the loss of the Hb A fraction as reference for interpretation.
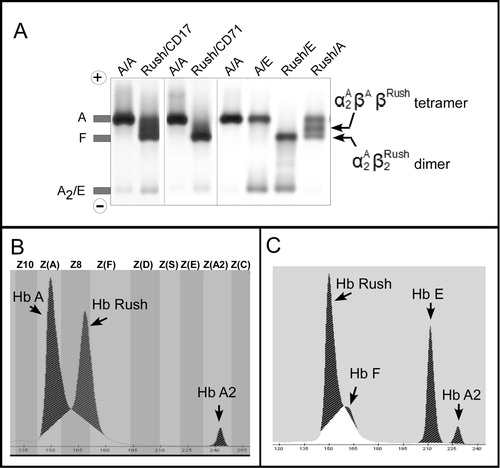
Sebia capillary 2 Flex-piercing electrophoresis in pH 9.4 condition provided the first evidence of a distinguishable Hb Rush fraction that migrated in the zone next to the Hb F fraction (B). No Hb F increase was observed in the Hb Rush simple heterozygote. The capillary electrophoresis revealed a single Hb Rush fraction, in contrast to the agarose electrophoresis, which revealed Hb Rush dimer and hybrid tetramer fractions (A). This result showed that the electrophoretic medium and pH conditions interfere with the separation profile of Hb Rush. We also observed that the capillary instrument could not automatically position and annotate hemoglobin fractions for the compound heterozygote of Hb Rush and Hb E owing to the absence of the Hb A fraction, a major reference required for zone interpretation of the software, in the compound heterozygote (C). Notably, the use of capillary electrophoresis revealed that Hb F production varied from normal to slightly elevated (1.4–3.7%) in the Hb Rush/Hb E compound heterozygotes. Unfortunately, Hb Rush/β0-thalassemia compound heterozygotes were not available for capillary electrophoresis analysis.
3.4. Hb Rush gene in the different Chinese families is linked to a common β-globin gene cluster haplotype
To investigate the origin of the Hb Rush mutation, we analyzed the β-globin gene cluster haplotypes of the studied families by using seven polymorphic restriction sites (). We found that Hb Rush variant was linked to a common haplotype [+----+-] in three unrelated families (families 1–3) with Thai and Han ancestry. Because the heterozygosity states of the polymorphic sites and the family members genotypes were unavailable, only the 5′ haplotype [+----], but not the 3′ haplotype (AvaII, Hinf I), was determined to be associated with the Hb Rush variant in the other 3 families (Families 4, 6 and 8). These results indicated that the βRush allele shares a common origin in the different Chinese ethnic families. In contrast, the βA allele was linked to at least 5 haplotypes with different 3′ subhaplotypes, thus indicating different origins of βA.
Table 2. Genotypes of polymorphic restriction sites of the β-globin gene cluster and the inferred haplotypes in the Chinese families from Southwest China.
4. Discussion
In the present study, we identified unstable Hb Rush variant in different Chinese ethnic families. We also revealed that Hb Rush in association with Hb E and β0 thalassemia lead to thalassemia intermedia with moderate anemia. At molecular level, we revealed hemoglobin electrophoresis patterns of the compound heterozygosity for Hb Rush and β thalassemia mutations. Moreover, we found the Hb Rush variant in the Chinese families with different ancestries was linked to a common β-globin gene cluster haplotype, thus suggesting a common origin of Hb Rush variant among different Chinese ethnic populations.
Unstable Hb Rush variant is a rare hemoglobin disorder previously described in only two unrelated families [Citation4,Citation5], and knowledge of its genotype-to-phenotype relationship is limited and unrecognized. The present study found that the most of Hb Rush simple heterozygote were asymptomatic, yet moderate anemia (HGB 85 g/L) was observed in one patient (subject P3 in ), suggesting that the clinical presentation of a simple heterozygous Hb Rush maybe heterogeneous. Variable hemoglobin concentrations were also observed in the same individual across multiple tests, thus reflecting the instability of the unstable Hb Rush. The mild hemolytic anemia of Hb Rush may be explained by its mild instability, which results from the substitution of a negatively charged glutamaic acid (Glu) by the neutral glutamine acid (Gln) in position 101 (G3) [Citation4,Citation13]. Although Heinz bodies are characteristic of unstable hemoglobins and hemolytic anemia [Citation14], they were observed in only a few members in an affected Brazilian family, and they were not found in the original study or our study. This suggests that the minor degree of Hb Rush instability generally does not cause an intracellular precipitation of hemoglobin in the form of Heinz bodies, and Heinz bodies may be overlooked. However, the clinical expression of the Hb Rush variant was heterogeneous and may be modified by a variety of environmental factors, such as smoking, exposure to oxidants and alterations of splenic functions [Citation15]. Therefore, the recognition and definition of this variant may be difficult. The severe anemia of Hb Rush may be induced by infection and fever, which may increase its instability by creating oxidative and heat stress conditions. Our study revealed the Hb Rush distribution in different Chinese populations in Southwest China, where infectious diseases are prevalent. The prevalence and clinical significance of Hb Rush are probably underestimated. It is important to establish appropriate screening strategies in these populations to prevent severe anemia associated with Hb Rush and guide proper treatments.
Our study is the first report to reveal the molecular and clinical consequences of unstable Hb Rush interacting with other hemoglobinopathies, and the results provide new insights into the genetic mechanisms responsible for β-thalassemia intermedia syndrome, which is a prevalent hemoglobin disorder exhibiting extreme genetic and clinical variability [Citation16]. Unstable hemoglobin interactions with β-thalassemia are one uncommon mechanism underlying thalassemia intermedia, only a few examples have been reported [Citation1,Citation17–19]. The oxygen binding properties and instability of unstable variants play roles in modifying the overall clinical expression of thalassemia intermedia. Highly unstable variants, such as Hb Genova, generally lead to severe thalassemia major syndrome [Citation2]. Compound heterozygosity for unstable Hb Rush and β-thalassemia traits including β0 and βE variants, produced thalassemia intermedia with moderate anemia in the present study. Hb Rush in the heterozygous state does not affect the interchain configurational change during oxygenation and displays a normal oxygen affinity [Citation4]. Therefore, the instability of Hb Rush appears to be the major determinant of variable anemia severity of thalassemia intermedia resulting from its association with β-thalassemia mutations.
The β-chain instability of Hb Rush that is responsible for hemolytic anemia may exacerbate the excess of α-globin chain exhibited in Hb E and β0-thalassemia carriers, and consequently aggravate the imbalance between α- and β-globin chains. The excess of free α-globin chains resulting from the thalassemia alleles may aggravate oxidative stress and thus increase the proportion of Hb Rush and its adverse effects. It is unlikely that heterozygous Hb Rush in association with the β-thalassemia traits produces more than a moderate hemolytic anemia. However, fever, oxidants and stress conditions can increase the instability of Hb Rush and therefore aggravated the anemia condition of the compound heterozygotes Hb Rush /Hb E and Hb Rush/β0-thalassemia. Our study provides new evidence and an example model of environmental influence on the clinical expression of thalassemia intermedia. Hb F induction was also observed in the Hb Rush compound heterozygotes, thus suggesting another possible modifying factor in the thalassemia intermedia [Citation20]. Our finding of Hb Rush interacting with respective β-thalassemia alleles extends the genetic spectrum and heterogeneity of thalassemia intermedia. Hemoglobin instability, environmental factors and Hb F are possible modifying factors that underlie the phenotypic heterogeneity of thalassemia intermedia associated with unstable Hb Rush.
Hemoglobin electrophoresis patterns of simple heterozygous Hb Rush have previously been reported, but patterns of compound heterozygous Hb Rush have not been characterized. As the Glu > Gln amino acid replacement results in a charge change, and Hb Rush was electrophoretically detectable in cellulose acetate and agarose gel electrophoreses in alkaline conditions. However, the migration of the Hb Rush faction is very similar to that of Hb F on these electrophoreses. As shown in our results, Hb Rush is distinguishable from Hb F on gel electrophoresis (A), thus potentially confounding diagnosis [Citation4,Citation5]. Our findings revealed the capillary electrophoresis pattern of Hb Rush, which showed clearly separation of Hb Rush from the Hb F fraction (B). Several Hb variants, such as Hb Atlanta, Hb Athens-GA (Waco) and Hb Alberta, exhibit very similar migration patterns to that of Hb Rush (HbVar database: http://globin.cse.psu.edu/hbvar/), but the distributions of these rare variants have not been observed in Southwest China. The present study firstly revealed the electrophoresis profiles of compound heterozygotes of Hb Rush with Hb E or β0-thalassemia mutations (), which is very informative for its screening and diagnosis. We speculate that the oxidative stress introduced by an excess of the α-chains due to β0-thalassemia and unstable Hb E exacerbated the instability of Hb Rush and increased its proportion. Therefore, Hb E and the β0-thalassemia may modify the clinical expression of unstable Hb Rush. The hemoglobin separation patterns demonstrated the molecular consequence of Hb Rush coinheriting with the other β-globin variants and explained the loss of normal hemoglobin function. Moreover, our findings suggest that molecular genetics methods are necessary for accurate screening and diagnosis of Hb Rush, as other abnormal hemoglobin variants could occur at β101 codon as well [Citation13,Citation21]. An Hpy188I restriction enzyme site is involved in the nucleotide substitution of Hb Rush, and a PCR-RFLP protocol may be used to detect the variant [Citation5]. The one-tube tetra-primer ARMS protocol we developed here have been proven as a rapid, cost-effective and accurate approach for screening of the Hb Rush variant.
Evolutionary information is useful for understanding the function and clinical implications of globin gene variants, De novo mutations and mutation hotspots may explain the origin of most unstable Hb variants, but the occurrence and origin of unstable Hb Rush variants had not been explored previously. There are four other hemoglobin variants discovered at the β101 position in addition to Hb Rush (Gln): Hb British Columbia (Lys), Hb Potomac (Asp), Hb Alberta (Gly) and the de novo mutation Hb Youngstown (Ala) [Citation13,Citation21]. The occurrence of these variants at the same codon suggests that the β101 codon is a hot-spot for mutations. However, our findings do not support a de novo mutation explaining the occurrence of the Hb Rush variant in the unrelated Chinese ethnic families. Our results demonstrated an autosomal dominant inheritance pattern of Hb Rush in the affected families. Moreover, the Hb Rush variant in the different Chinese families was linked to a common β-globin gene cluster haplotype [+----+-], thus suggesting a common origin of the variant in the populations of Southwest China, presumably owing to a founder effect. However, the haplotype [+----+-] is a prevalent haplotype that is associated with thalassemia and normal β-globins in the region [Citation22], it may be generated by recombination and gene conversion events [Citation23]. Therefore, our findings do not exclude the possibility that the unstable Hb Rush variant might have arisen more than once in Chinese populations. The Chinese families in this study and the two previously reported families of African and European origins exhibited wide geographical isolation and significant race differences [Citation4,Citation5]. Therefore, we propose that the Hb Rush variant has experienced multiple independent origins worldwide.
In summary, this study characterized the genetic and hematological features of Hb Rush interactions with thalassemia mutations, which would provide an interesting model to gain insights into the possible mechanisms of the gene-gene interactions of different globin genes and the genetic heterogeneity underlying the clinical heterogeneity of thalassemia. The instability of unstable Hb Rush mediates environmental influences on phenotype, thus providing new insights into the contribution of environmental modifying factors to the complexity of inherited blood disorders. This study demonstrated that Hb Rush is distinguishable from other Hb variants with innocuous phenotypes in the region, thus suggesting the necessity of Hb Rush screening and genetic counseling in the region. However, the present study also has limitations, including the small numbers of pedigrees and patients for evaluating the function of various Hb Rush heterozygotes. Further investigation on the distribution, origin and genotype-to-phenotype relationship of unstable hemoglobin Rush and its interaction with other hemoglobinopathies would greatly improve the diagnosis, treatment and control of hemoglobinopathies.
Supplemental Material
Download MS Word (1.8 MB)Acknowledgement
This work was supported by the Applied Basic Research Foundation of Yunnan Province (No. 2016FA048 and 2018FE001-049) and a grant from the National Key Research and Development Program of China (No. 2016YFC1201704)
Disclosure statement
No potential conflict of interest was reported by the authors.
ORCID
Additional information
Funding
Related Research Data
References
- Papassotiriou I, Traeger-Synodinos J, Prome D, et al. Association of unstable hemoglobin variants and heterozygous beta-thalassemia: example of a new variant Hb Acharnes or [beta53(D4) Ala –> Thr]. Am J Hematol. 1999;62(3):186–192. doi: 10.1002/(SICI)1096-8652(199911)62:3<186::AID-AJH9>3.0.CO;2-Z
- Badens C, Paolasso C, Fossat C, et al. Compound heterozygosity for unstable hemoglobin Genova and beta(o)-thalassemia associated with early onset of thalassemia major syndrome. Haematologica. 2005;90(1):ECR04.
- Panigrahi I, Marwaha RK, Kulkarni K. The expanding spectrum of thalassemia intermedia. Hematology. 2009;14(6):311–314. doi: 10.1179/102453309X12473408860307
- Adams JG, 3rd, Winter WP, Tausk K, et al. Hemoglobin Rush (beta 101 (g3) glutamine): a new unstable hemoglobin causing mild hemolytic anemia. Blood. 1974;43(2):261–269.
- Silva MR, Sendin SM, Velloso-Rodrigues C, et al. Unstable hemoglobin Rush [beta 101(G3) Glu>Gln, HBB:c.304G>C] in a Brazilian family with moderate hemolytic anemia. Ann Hematol. 2012;91(7):1091–1096. doi: 10.1007/s00277-011-1403-0
- Zhang J, He J, Zeng XH, et al. Genetic heterogeneity of the beta-globin gene in various geographic populations of Yunnan in southwestern China. PLoS One. 2015;10(4):e0122956. doi: 10.1371/journal.pone.0122956
- Liu H, Huang K, Liu S, et al. Gene frequency and haplotype distribution of hemoglobin E among seven minority groups of Yunnan, China. Am J Hum Biol. 2016;28(6):927–931. doi: 10.1002/ajhb.22868
- Tan AS, Quah TC, Low PS, et al. A rapid and reliable 7-deletion multiplex polymerase chain reaction assay for alpha-thalassemia. Blood. 2001;98(1):250–251. doi: 10.1182/blood.V98.1.250
- Wee YC, Tan KL, Chua KH, et al. Molecular characterisation of Haemoglobin Constant Spring and Haemoglobin Quong Sze with a combine-amplification refractory mutation system. Malays J Med Sci. 2009;16(3):23–30.
- Medrano RF, De Oliveira CA. Guidelines for the tetra-primer ARMS-PCR technique development. Mol Biotechnol. 2014;56(7):599–608.
- Lee YJ, Park SS, Kim JY, et al. RFLP haplotypes of beta-globin gene complex of beta-thalassemic chromosomes in Koreans. J Korean Med Sci. 2002;17(4):475–478. doi: 10.3346/jkms.2002.17.4.475
- Varawalla NY, Fitches AC, Old JM. Analysis of beta-globin gene haplotypes in Asian Indians: origin and spread of beta-thalassaemia on the Indian subcontinent. Hum Genet. 1992;90(4):443–449. doi: 10.1007/BF00220475
- Shih DT, Jones RT, Imai K, et al. Involvement of Glu G3(101)beta in the function of hemoglobin. Comparative O2 equilibrium studies of human mutant hemoglobins. J Biol Chem. 1985;260(10):5919–5924.
- Williamson D. The unstable haemoglobins. Blood Rev. 1993;7(3):146–163. doi: 10.1016/0268-960X(93)90002-L
- Zinkham WH, Winslow RM. Unstable hemoglobins: influence of environment on phenotypic expression of a genetic disorder. Medicine (Baltimore). 1989;68(5):309–320. doi: 10.1097/00005792-198909000-00005
- Asadov C, Alimirzoeva Z, Mammadova T, et al. Beta-Thalassemia intermedia: a comprehensive overview and novel approaches. Int J Hematol. 2018;108(1):5–21. doi: 10.1007/s12185-018-2411-9
- Galacteros F, Loukopoulos D, Fessas P, et al. Hemoglobin Koln occurring in association with a beta zero thalassemia: hematologic and functional consequences. Blood. 1989;74(1):496–500.
- Vassilopoulos G, Papassotiriou I, Voskaridou E, et al. Hb Arta [beta 45 (CD4) Phe–>Cys]: a new unstable haemoglobin with reduced oxygen affinity in trans with beta-thalassaemia. Br J Haematol. 1995;91(3):595–601. doi: 10.1111/j.1365-2141.1995.tb05353.x
- Bardakdjian-Michau J, Fucharoen S, Delanoe-Garin J, et al. Hemoglobin Dhonburi alpha 2 beta 2 126 (H4) Val----Gly: a new unstable beta variant producing a beta-thalassemia intermedia phenotype in association with beta zero-thalassemia. Am J Hematol. 1990;35(2):96–99. doi: 10.1002/ajh.2830350206
- Thein SL. Genetic basis and genetic modifiers of beta-thalassemia and sickle cell disease. Adv Exp Med Biol. 2017;1013:27–57. doi: 10.1007/978-1-4939-7299-9_2
- Edward HL, Pisani LA, Rodriguez-Romero WE, et al. Hb Youngstown [beta101(G3)Glu –> Ala; HBB: c.305A > C]: an unstable hemoglobin variant causing severe hemolytic anemia. Hemoglobin. 2014;38(6):381–384. doi: 10.3109/03630269.2014.971960
- Sun H, Liu H, Huang K, et al. Beta-globin gene cluster haplotypes in ethnic minority populations of southwest China. Sci Rep. 2017;7:42909), doi: 10.1038/srep42909
- Long JC, Chakravarti A, Boehm CD, et al. Phylogeny of human beta-globin haplotypes and its implications for recent human evolution. Am J Phys Anthropol. 1990;81(1):113–130. doi: 10.1002/ajpa.1330810112