
ABSTRACT
Objectives: Acute myeloid leukemia (AML) is a heterogeneous and highly recurrent hematological malignancy. Studies have shown an association between microRNAs and drive genes in AMLs. However, the regulatory roles of miRNAs in AML and how they act on downstream targets and the signaling pathway has been little studied.
Methods: As to understand the mechanism of mRNA-miRNA interaction in the blood malignancy from a large scale of transcriptomic sequencing studies, we applied a comprehensive miRNA-mRNA association, co-expression gene network and ingenuity pathway analysis using TCGA AML datasets.
Results: Our results showed that his-mir-335 was a critical regulatory of homeobox A gene family. PBX3, KAT6A, MEIS1, and COMMD3-BMI1 were predicted as top transcription regulators in the regulatory network of the HOXA family. The most significantly enriched functions were cell growth, proliferation, and survival in the mRNA-miRNA network.
Conclusion: Our work revealed that regulation of the HOXA gene family and its regulation played an important role in the development of AML.
Introduction
Acute myeloid leukemia (AML), is the most common hematological malignancy characterized by the abnormal proliferation and differentiation of myeloid progenitor cells with high incidence and recurrence rates [Citation1]. AML is shown to be a type of disorder in the clonal form but is also relevant heterogeneous concerning the genotypic, phenotypic, and clinical characteristics.
With the development of sequencing techniques, targeted therapy facilitated by molecular biology and genetics is a promising area for therapy of AML [Citation2]. High throughput technologies such as RNA sequencing have enabled the revelation of global transcriptomic changes in AML, especially key molecules in the pathogenesis. Currently, studies indicated that during the pathogenesis of AML regulation of a variety of signaling pathways were overexpressed which was caused by mutations contributing to hematopoietic transformation in terms of transcriptional targets during pathogenesis, such as TET2, DNMT3A, ASXL1, IDH1 and IDH2 [Citation3]. The different signaling pathways are also involved in the pathogenesis of AML including PI3K/AKT pathway, MLL B-ALL gene expression signatures, and HOX gene family
MicroRNAs (miRNAs) are a group of conserved short non-coding RNAs (19–25nt) [Citation4] that regulate mRNA through targeting their seed sequences [Citation5], playing crucial roles in biological functions during hematopoiesis and apoptosis in blood cells [Citation6]. Although previous studies showed that miR-126, miR-196b, miR-10b, miR-21 played important roles in mutated FLT3-ITD, RUNX1, RUNX1-RUNX1T1, and CBFB-MYH11 associated AMLs, understanding of the distinctive pattern of miRNAs is still not enough to thoroughly reveal the mechanism of AML. A combined microRNA and mRNA analysis to deeper understand the pathogenesis of AML is needed.
The Cancer Genome Atlas (TCGA) project is aimed to profile genomic changes in more than 33 different cancer types, including AML, through a better understanding of the genetic basis of the disease. In this study, we investigated the TCGA transcriptomic dataset of AML to reveal the regulatory events between miRNA and mRNA, which were related to AML. Herein, as to comprehensively understand the mechanism of mRNA-miRNA interaction in the blood malignancy, we applied the TCGA AML dataset for miRNA-mRNA association analysis, co-expression gene network analysis, and pathway analysis.
Methods
Data description and preprocess
The sequencing-based mRNA and miRNA expression data of 179 AML patients were obtained from the TCGA project. The gene expression FPKM (fragment per kilobase per million mapped reads) values were calculated. Gene with a zero FPKM value in all samples was discarded. After normalization 17,300 mRNAs and 612 miRNAs remained. To avoid infinite values, a value of 0.001 was added to the FPKM value before log2 transformed for each gene.
miRNA-mRNA association analysis
The pair-wise Pearson correlation coefficient between mRNA and miRNA expression was calculated based on 17,300 mRNAs and 612 miRNAs, yielding a correlation coefficient data matrix with 17,300 mRNAs in a row and 612 miRNAs in columns. A Pearson Correlation Coefficient less than −0.6 was used as a cutoff to obtain the most probable biologically relevant miRNA-mRNA regulations. We only select the strongest negative correlations (−0.6 or less) to draw in miRNA-mRNA regulations network graphs with Cytoscape. Data preprocessing, the Pearson correlation coefficient was performed in the R (www.r-project.org/) environment with its ‘base’ function and ‘stat’ packages.
Integrity pathway analysis of target mRNA
Target mRNA identified by miRNA-mRNA association analysis (cor < −0.6) was used for functional annotation and pathway analysis to determine the biological function of these genes. The enriched pathway was determined by both significant fisher exact test (p-value < 0.05), and at least three genes were involved in the pathway.
Results
Correlation analysis identified the miRNA-mRNA interaction network in AML
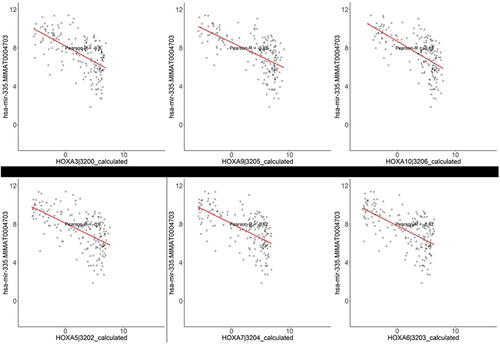
The microRNAs are regulatory, play an important role in many cancer types which may be involved in cancer initiation and development. To identify miRNA associated with AML, we download transcriptome sequencing data of AML, including both mRNA and miRNA gene expression profiles of 179 tumor samples. Given the regulatory relationship between miRNA and mRNA, we assumed that the existence of a negative correlation between miRNA and the expression of its target genes. By performing correlation analysis, we obtained a group of miRNA-mRNA pairs. We applied a cutoff of negative regulation with a correlation coefficient <−0.6 to select the most significant miRNA association pairs, which might be strongly associated with the initiation and progression of AML. The network in shows the miRNA-mRNA interaction network in AML. We observed that several miRNA functions as hub regulators in the network, such as has-mir-335, has-mir-146a, and has-mir-21. These miRNA have been reported to be up-regulated in AML, but the clear mechanism of the regulatory in cancer remains unknown. Notably, the expression of has-mir-135 in AML was associated with five family members of homeobox genes. As shown in , we observed that expression of has-mir-335 was significantly associated with HOXA with correlation <−0.6, which might indicate has-mir-335 targeted the HOXA family members.
Figure 1. miRNA-mRNA regulation network identified by gene-expression correlation analysis.

Figure 2. Correlation between expression of has-mir-335 and HOXA family members. Each dot represents a patient.
Function annotation for miRNA regulated mRNA in AML
To further characterize the role of miRNA and their target mRNA in AML, we perform a function annotation for all the mRNA in the regulation network. As shown in , most of the mRNA in the network of is involved in cell growth, proliferation, and survival.
Table 1. IPA function annotation for Targeted mRNAs in the regulation network.
We also perform an IPA pathway analysis by using these target mRNA. The enriched pathways were listed in . Salvage Pathways of Pyrimidine Deoxyribonucleotides was the top pathway with a −log (p-value) 2.63, ATP and TK2 gene in the regulation network that was involved in this pathway. Previous studies [Citation7,Citation8] have demonstrated the important role of deoxyribonucleotides metabolism in leukemia and other cancer types. Besides, Glutamyl Cycle and Colanic Acid Building Blocks Biosynthesis were also significantly enriched in leukemia. Notably, we observed that ATP was involved in most enriched pathways. It was indicated ATP associated with energy metabolism might play an important role in AML.
Table 2. IPA pathway analysis for Targeted mRNAs in the regulation network.
Gene-expression regulator prediction for Targeted mRNAs in the regulation network
We also use IPA to predict the potential regulator of these targeted genes. As shown in , PBX3 was predicted as a top transcription regulator of six target genes in the regulatory network HOXA10, HOXA3, HOXA5, HOXA6, HOXA7, and HOXA9. HOXA and PBX3 are independent predictors of poor survival in patients with acute myeloid leukemia [Citation9,Citation10] and also associated with the chemotherapy of AML. Except for PBX3, there are also 12 genes that were predicted as a transcription regulator of several family members of homeobox A, including the KAT6A gene which was reported as a commonly rearranged gene in AML [Citation11]. Our results suggested regulation of HOXA gene expression might be associated with the development of AML.
Table 3. IPA gene-expression regulator prediction for Targeted mRNAs in the regulation network.
Discussion
In this study, we investigated a transcriptome dataset of AML to reveal the regulatory events between miRNA and mRNA, which were related to AML. Our results showed that his-mir-335 was a key regulatory of homeobox A gene family. Regulation of the HOXA gene family may play an important role in the development of AML. Studies have demonstrated that genetic alterations accumulation might contribute to the initiation of AML. Despite an increased understanding of AML, our knowledge about transcriptome regulation in AML still needed to be improved.
Firstly, our study predicted a series of miRNA regulators in AML by performing correlation analysis between miRNA and mRNA, including has-mir-130a, has-mir-146a, has-mir-335, has- has-mir-21, has-mir-126, and has-mir-181a/b/c/d. Previous studies had revealed these miRNAs played important roles in the development of AML. Has-mir-130a was involved in the megakaryocytic differentiation and erythropoiesis, and its expression was also related to the differentiation patterns in AML [Citation12]. Has-mir-146a was a regulator of apoptosis [Citation13], and its expression was associated with patient prognosis of AML. The increase of has-mir-146a can enhance the sensitivity of leukemic blast cells to cytotoxic drugs [Citation14]. Besides, has-mir-335, has-mir-21, and has-mir-126 were significantly higher expressed. Expression of has-mir-335 was associated with patient clinical outcomes [Citation15]. Has-mir-181 family members were reported to be associated with the myeloid differentiation, and has-mir-181a could enhance cell proliferation [Citation16] and correlated with the morphological sub-type of AML [Citation17]. However, research also demonstrated that has-mir-181a acts as a tumor suppressor in the pathogenesis of AML [Citation18]. Our study provided further evidence for the regulatory role of these miRNAs in AML.
Next, we observed that the target genes of hub miRNAs were related to cell growth, proliferation, death, and survival. Pathway analysis demonstrated that the ATP enzyme was significantly affected by miRNA regulation. The ATP was involved in biosynthesis pathways which were significantly enriched. Salvage Pathways of Pyrimidine Deoxyribonucleotides was a pathway related to the biosynthesis of deoxyribonucleotides. ATP played a very important role in this biological procedure [Citation19–23]. Deoxyribonucleotides were essential to DNA synthesis/repair. The previous study demonstrated that the biosynthesis of deoxyribonucleotides was an important target for anticancer therapy [Citation24,Citation25].
Finally, we reveal that expression six homeobox gene family members were related to has-mir-335. The dysregulation of HOX genes is associated with AML [Citation26]. Recent work has shown that their overexpression is important in the formation of malignancy [Citation27,Citation28]. Our regulatory prediction results showed that PBX3 was a key regulatory of HOXA genes. Homeobox family, including four members (A, B, C, and D) along with 39 homeotic or Hox genes, are conserved and are critical in regulating differentiation or proliferation [Citation9]. PBX3 was an important cofactor of HOXA9 in leukemogenesis [Citation9]. Our study suggests that regulation between has-mir-335 and HOXAs plays an important role in AML, which provided a novel insight for AML researches in terms of transcriptome regulation.
Data availability
The sequencing-based mRNA and miRNA expression data of 179 AML patients were obtained from the TCGA project (http://cancergenome.nih.gov/).
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Slusher TM, Olusanya BO, Vreman HJ, et al. A randomized trial of phototherapy with filtered sunlight in African neonates. N Engl J Med. 2015;373:1115–1124. doi: 10.1056/NEJMoa1501074
- Trino S, Lamorte D, Caivano A, et al. MicroRNAs as new biomarkers for diagnosis and prognosis, and as potential therapeutic targets in acute myeloid leukemia. Int J Mol Sci. 2018;19:460. doi: 10.3390/ijms19020460
- Shih AH, Abdel-Wahab O, Patel JP, et al. The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer. 2012;12:599–612. doi: 10.1038/nrc3343
- Vitsios DM, Davis MP, van Dongen S, et al. Large-scale analysis of microRNA expression, epi-transcriptomic features and biogenesis. Nucleic Acids Res. 2017;45:1079–1090. doi: 10.1093/nar/gkw1031
- Wallace JA, O'Connell RM. MicroRNAs and acute myeloid leukemia: therapeutic implications and emerging concepts. Blood. 2017;130:1290–1301. doi: 10.1182/blood-2016-10-697698
- Pichiorri F, De Luca L, Aqeilan RI. MicroRNAs: new players in multiple myeloma. Front Genet. 2011;2:22. doi: 10.3389/fgene.2011.00022
- Mathews CK. Deoxyribonucleotide metabolism, mutagenesis and cancer. Nat Rev Cancer. 2015;15:528–539. doi: 10.1038/nrc3981
- Sylwestrowicz T, Piga A, Murphy P, et al. The effect of deoxycoformycin and deoxyadenosine on deoxyribonucleotide concentrations in leukaemic cells. Br J Haematol. 1982;51:623–630. doi: 10.1111/j.1365-2141.1982.tb02826.x
- Li Z, Zhang Z, Li Y, et al. PBX3 is an important cofactor of HOXA9 in leukemogenesis. Blood. 2013;121:1422–1431. doi: 10.1182/blood-2012-07-442004
- Dickson GJ, Liberante FG, Kettyle LM, et al. HOXA/PBX3 knockdown impairs growth and sensitizes cytogenetically normal acute myeloid leukemia cells to chemotherapy. Haematologica. 2013;98:1216–1225. doi: 10.3324/haematol.2012.079012
- Chinen Y, Taki T, Tsutsumi Y, et al. The leucine twenty homeobox (LEUTX) gene, which lacks a histone acetyltransferase domain, is fused to KAT6A in therapy-related acute myeloid leukemia with t(8;19)(p11;q13). Genes Chromosomes Cancer. 2014;53:299–308. doi: 10.1002/gcc.22140
- Garzon R, Croce CM. MicroRNAs in normal and malignant hematopoiesis. Curr Opin Hematol. 2008;15:352–358. doi: 10.1097/MOH.0b013e328303e15d
- Yan W, Guo H, Suo F, et al. The effect of miR-146a on STAT1 expression and apoptosis in acute lymphoblastic leukemia Jurkat cells. Oncol Lett. 2017;13:151–154. doi: 10.3892/ol.2016.5395
- Spinello I, Quaranta MT, Riccioni R, et al. MicroRNA-146a and AMD3100, two ways to control CXCR4 expression in acute myeloid leukemias. Blood Cancer J. 2011;1:e26. doi: 10.1038/bcj.2011.24
- Lin X, Wang Z, Zhang R, et al. High serum microRNA-335 level predicts aggressive tumor progression and unfavorable prognosis in pediatric acute myeloid leukemia. Clin Transl Oncol. 2015;17:358–364. doi: 10.1007/s12094-014-1237-z
- Verduci L, Azzalin G, Gioiosa S, et al. microRNA-181a enhances cell proliferation in acute lymphoblastic leukemia by targeting EGR1. Leuk Res. 2015;39:479–485. doi: 10.1016/j.leukres.2015.01.010
- Su R, Lin HS, Zhang XH, et al. MiR-181 family: regulators of myeloid differentiation and acute myeloid leukemia as well as potential therapeutic targets. Oncogene. 2015;34:3226–3239. doi: 10.1038/onc.2014.274
- Weng H, Lal K, Yang FF, et al. The pathological role and prognostic impact of miR-181 in acute myeloid leukemia. Cancer Genet. 2015;208:225–229. doi: 10.1016/j.cancergen.2014.12.006
- Dance GS, Beemiller P, Yang Y, et al. Identification of the yeast cytidine deaminase CDD1 as an orphan C–>U RNA editase. Nucleic Acids Res. 2001;29:1772–1780. doi: 10.1093/nar/29.8.1772
- Kern L, de Montigny J, Jund R, et al. The FUR1 gene of Saccharomyces cerevisiae: cloning, structure and expression of wild-type and mutant alleles. Gene. 1990;88:149–157. doi: 10.1016/0378-1119(90)90026-N
- Kurtz JE, Exinger F, Erbs P, et al. The URH1 uridine ribohydrolase of Saccharomyces cerevisiae. Curr Genet. 2002;41:132–141. doi: 10.1007/s00294-002-0296-9
- Kurtz JE, Exinger F, Erbs P, et al. New insights into the pyrimidine salvage pathway of Saccharomyces cerevisiae: requirement of six genes for cytidine metabolism. Curr Genet. 1999;36:130–136. doi: 10.1007/s002940050482
- Mitterbauer R, Weindorfer H, Safaie N, et al. A sensitive and inexpensive yeast bioassay for the mycotoxin zearalenone and other compounds with estrogenic activity. Appl Environ Microbiol. 2003;69:805–811. doi: 10.1128/AEM.69.2.805-811.2003
- Shao J, Zhou B, Chu B, et al. Ribonucleotide reductase inhibitors and future drug design. Curr Cancer Drug Targets. 2006;6:409–431. doi: 10.2174/156800906777723949
- Aimiuwu J, Wang H, Chen P, et al. RNA-dependent inhibition of ribonucleotide reductase is a major pathway for 5-azacytidine activity in acute myeloid leukemia. Blood. 2012;119:5229–5238. doi: 10.1182/blood-2011-11-382226
- Alharbi RA, Pettengell R, Pandha HS, et al. The role of HOX genes in normal hematopoiesis and acute leukemia. Leukemia. 2013;27:1000–1008. doi: 10.1038/leu.2012.356
- Ferrando AA, Armstrong SA, Neuberg DS, et al. Gene expression signatures in MLL-rearranged T-lineage and B-precursor acute leukemias: dominance of HOX dysregulation. Blood. 2003;102:262–268. doi: 10.1182/blood-2002-10-3221
- Rozovskaia T, Feinstein E, Mor O, et al. Upregulation of Meis1 and HoxA9 in acute lymphocytic leukemias with the t(4 : 11) abnormality. Oncogene. 2001;20:874–878. doi: 10.1038/sj.onc.1204174