
ABSTRACT
Objectives: Hereditary spherocytosis (HS) represents a group of congenital diseases characterized by sphere-shaped erythrocytes on peripheral blood smears. The typical clinical manifestations of HS include haemolysis, jaundice, splenomegaly, and gallstones. Ankyrin1, encoded by the ANK1 gene, is the predominant protein in red blood cells. Defects in ankyrin1 lead to a decrease in erythrocyte surface area, a spherical shape of erythrocytes and, in particular, loss of membrane elasticity and mechanical stability. The purpose of this study was to investigate a Chinese family with HS to determine the causative gene mutation and explore the genotype–phenotype correlation.
Methods: A 4-year-old boy was diagnosed with HS based on typical clinical features. In addition, his father had a high possibility of HS. Targeted next-generation sequencing (NGS) followed by Sanger sequencing was performed in the proband and his parents.
Results: One mutation in the ANK1 gene was recognized. c1801-1G > C in exon 17, which leads to splicing defects, was detected. To confirm the c1801-1G > C variant, samples from the proband and his parents were analysed by Sanger sequencing, and Sanger verification showed that this mutation was inherited from the father.
Conclusion: The present study confirmed that a novel mutation in ANK1 may be causative of HS, which plays an important role in expanding the mutational spectrum of ANK1 mutations. This may contribute to accurate genetic counselling. And it is helpful for understanding the correlation of the genotype and phenotype.
Introduction
Hereditary spherocytosis (HS) is the most common cause of haemolytic anaemia due to erythrocyte membrane defects, but only one-third of the affected infants are diagnosed with HS in the first year of life [Citation1]. HS can be observed in all populations, although Caucasians have the highest incidence [Citation2]. Among Chinese populations, the incidence of HS is approximately 1/100000 [Citation3]. Typical clinical manifestations of HS include anaemia, haemolysis, jaundice, splenomegaly and gallstones [Citation4]. However, clinical presentations vary widely, ranging from nearly asymptomatic to transfusion-dependent or severe life-threatening anaemia, and can be aggravated by infection, pregnancy, and sudden haemorrhage, among others [Citation5]. In the neonatal period, jaundice and anaemia are the most common clinical features. Splenomegaly is rarely detected in neonates, and spherocytes are less often observed on blood smears of neonates. Furthermore, biochemical tests, such as osmotic fragility and eosin-5′-maleimide, are unreliable due to the lack of age-appropriate controls [Citation6]. Therefore, the diagnosis of HS in neonates can be challenging [Citation5,Citation6]. Treatment of HS mainly includes blood transfusions and splenectomy to alleviate haemolysis and clinical symptoms.
To date, five HS-related genes have been identified, namely, ANK1 (ankyrin1), SPTB (β-spectrin), SPTA1 (α-spectrin), SLC4A1 (band 3), and EPB42 (protein 4.2) [Citation7]. Mutations in ANK1 are the main cause of HS. Most of these mutations are nonsense mutations, frameshift mutations or splicing mutations [Citation8]. ANK1 is composed of 42 exons encoding a 1881-amino acid protein. ANK1 consists of an N-terminal domain containing multiple ankyrin repeats, a central region with a spectrin-binding domain, and a C-terminal regulatory domain that is the least conserved and variable [Citation9].
Although autosomal recessive inheritance and de novo mutations have also been described, most HS cases are display autosomal dominance [Citation10]. In cases with a known family history of HS and the presence of typical clinical and biological features, it is sufficient to make the diagnosis by a confirmatory test. However, in cases with a negative family history or with atypical features, molecular genetic analysis can help to make the diagnosis and determine the genetic pattern [Citation11].
In this case report, a heterozygous ANK1 c.1801-1G > C mutation was identified by targeted next-generation sequencing (NGS) and Sanger sequencing in two patients from a Chinese family. The genetic characteristics and clinical features are presented in detail.
Case presentation
A 4-year-old boy with fever, anaemia, and jaundice was referred to our centre. He had fever for five days before being referred to our department. Pale lips and jaundice were observed soon after he was admitted to the hospital. Hepatomegaly and splenomegaly were identified by physical examination. The boy had a medical history of mild anaemia of unknown cause, of approximately 90 g/L–100 g/L, since infancy. His anaemia and jaundice had become rapidly worse during the past few days. The results of blood tests at the time of admission to our hospital are shown in , which indicated that the boy suffered from haemolytic anaemia and hyperbilirubinemia. No abnormalities were observed by glucose-6-phosphate dehydrogenase (G6PD) activity analysis, direct Coombs test, or haemoglobin electrophoresis. Erythrocyte osmotic fragility was increased. Densely stained spherical red blood cells (RBC) were frequently observed in the peripheral blood smear (A). Then, the proband was clinically diagnosed with HS. The proband’s father had shown similar symptoms, such as anaemia and jaundice, since he was young. A haematological investigation of the proband’s father showed the possibility of compensated haemolysis (). Splenomegaly was detected by B-ultrasound. Clinical manifestations and haematological data of the proband’s father indicated a high possibility of HS ().
Figure 1. Heterozygous ANK1 c.1801-1G > C mutation in two patients from a Chinese family with hereditary spherocytosis. A. Spherocytes were observed in the film of the proband. B. Family tree and the genotype at the ANK1 c.1801-1 position. Squares and circles denote males and females, respectively. Black symbols denote patients with HS.
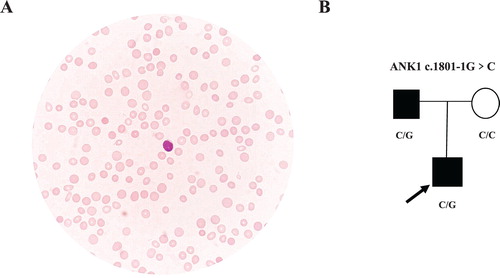
Figure 2. Sequencing diagram of family members. Sequencing diagrams indicate the presence of the ANK1 mutation in the proband and his father but not in his mother.
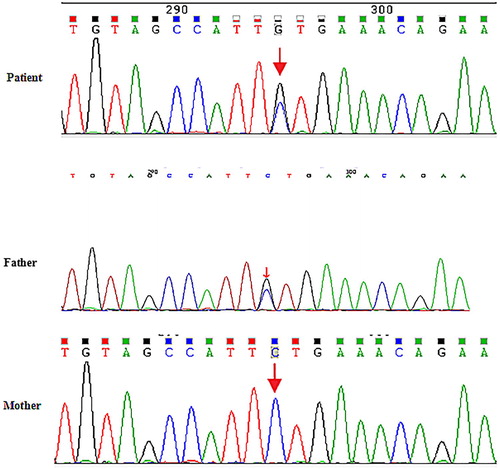
Table 1. Laboratory test results of the patient and his father.
Methods
To confirm the genetic cause of HS quickly, a total of 208 genes were selected as candidates that may lead to the pathogenesis of hereditary erythroid-related diseases using the GenCap custom enrichment kit (MyGenostics Inc., Beijing, China).
Genomic DNA was extracted using the QIAampBlood Midi Kit (QIAGEN, Valencia, CA) from the peripheral blood leukocytes of the patient and his family members. DNA libraries were prepared according to Illumina protocol. The enriched libraries were sequenced on an Illumina NextSeq 500 sequencer (Illumina, San Diego, CA, USA) for 150 bp paired-end reads. Following sequencing, low-quality variations were filtered out using a quality score ≥20, and BWA was used to align the clean reads to the reference human genome (hg19). Local realignment around indels and base quality score recombination by GATK were performed on the BAM file for pre-processing. Unified Genotyper and Hapolotype Caller in GATK (http://www.broadinstitute.org/gsa/wiki/index.php/Home_Page) and SOAPsnp program (http://soap.genomics.org.cn/soapsnp.html) were used to call variants. Rare mutations, whose population frequencies were lower than 1%, were filtered according to the 1000 Genomes data, ESP6500 population data, and ExAC data. The pathogenicity of other rare mutations was annotated by SIFT (http://sift.bii.a-star.edu.sg/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and MutationTaster (http://www.mutationtaster.org/). Variants identified with target next-generation sequencing were confirmed by Sanger sequencing (Figure 2).
Results
In this family, the patient was diagnosed with HS based on his typical clinical features. One mutation in the ANK1 gene was recognized. The c1801-1G > C mutation in exon 17 leads to splicing defects. This mutation is not found in HapMap samples, 1000 Genomes, ESP6500, ExAC_ALL or ExAC_EAS population databases. Predictions by FATHMM and MutationTaster revealed that the mutation was deleterious, and GEREP++ predicted that the mutation was in a conserved region. To confirm the c1801-1G > C variant, we analysed samples from the patient and his parents by Sanger sequencing, and Sanger verification showed that this mutation was inherited from the father (B-2).
Discussion
HS represents a group of congenital diseases characterized by the presence of spherical erythrocytes in peripheral blood smears [Citation12]. Moreover, HS is the most common cause of congenital haemolytic anaemia due to membrane protein defects [Citation13]. The ANK1 gene is located on chromosome 8p11.1 and encodes several alternatively spliced isoforms [Citation14]. Erythroid ankyrin is the predominant protein encoded by ANK1 in red blood cells. Because ankyrin attaches β-spectrin to the band 3 protein, lack of ankyrin leads to a proportional and secondary decrease in spectrin assembly on the membrane, in spite of normal spectrin synthesis [Citation15]. Defects in these proteins lead to a decrease in the erythrocyte surface area, a spherical shape of erythrocytes and, in particular, loss of membrane elasticity and mechanical stability [Citation16].
In this report, we described a 4-year-old Chinese boy with typical presentations of anaemia, jaundice and splenomegaly. He had been suffering from mild anaemia since birth. His anaemia became aggravated with infection. Spherocytes were observed in his peripheral blood smears. Family history revealed that the father had similar symptoms, including anaemia, jaundice and splenomegaly, in his childhood. The proband’s father gradually recovered with age. A gene panel including 208 genes related to hereditary erythroid-related diseases identified a novel heterozygous mutation c.1801-1G > C on ANK1 in the proband, which had been inherited from his father. The ANK1 variant detected by NGS was confirmed by Sanger sequencing.
Ankyrin mutations are the most common cause of HS in Northern European populations and were first reported in German patients [Citation1]. In recent years, several new pathogenic mutations have been identified relative to ANK1-mutated HS in some Asian countries, such as China, Japan and Korea [Citation17]. Thus far, over 80 different mutations have been confirmed in ANK1-mutated HS patients. Homozygous mutations have not yet been described; however, it is uncertain whether mild homozygous or compound homozygous mutations will mimic de novo disease [Citation18]. Frameshift mutations and nonsense mutations in the ANK1 gene are mainly detected in patients with HS displaying autosomal dominant (AD) inheritance, and point mutations (missense mutations) are mainly detected in patients who display non-AD transmission [Citation1].
HS can be diagnosed at any age and with any severity, and the description of case reports varies greatly. Luo et al. identified a novel splice-site ANK1 mutation (c.2960 + 2T > G) in newborns based on significantly decompensated anaemia and unexplained jaundice in early infancy, family history, and haematological data [Citation17]. In general, patients with HS may have a severe phenotype early in life, and the symptoms may disappear with age. However, severe phenotypes in adults have also been reported. A 65-year-old female was referred to a Korean hospital due to haemolytic anaemia. She received red blood cell (RBC) transfusion several times and cholecystectomy approximately 20 years ago. Abundant densely staining spherical-shaped erythrocytes (spherocytes) are found on peripheral blood smears. A novel nonsense heterozygous mutation (c.1956G > A) on SPTB was identified by NGS. This female underwent splenectomy due to transfusion-refractory anaemia and splenomegaly [Citation19].
Splenectomy is the only curative treatment available for HS at present, and the appropriate age for splenectomy is 5 years or more [Citation20]. Splenectomy can only eliminate haemolysis and associated signs and symptoms but cannot correct cytoskeletal membrane defects in HS [Citation21]. In our study, the patient received RBC transfusion while in the hospital. After infection control, he partially recovered with a haemoglobin level of 90–100 g/L. The growth and development of the patient are normal, and thus, we are paying close attention to changes following his discharge from the hospital.
In summary, a novel splicing mutation in ANK1 was identified using NGS in a Chinese family affected by HS. Identifying potential genetic causes is helpful not only in accurate genetic counselling but also understanding the correlation of the genotype and phenotype. NGS provides a useful diagnostic tool for the molecular diagnosis of HS, especially in atypical cases with no family history or in cases of negative results on traditional diagnostic tests.
Acknowledgement
We thank all patients for their participation.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- Eber SW, Gonzale JM, Lux ML, et al. Ankyrin-1 mutations are a major cause of dominant and recessive hereditary spherocytosis. Nat Genet. 1996;13(2):214–218. doi: 10.1038/ng0696-214
- Perrotta S, Gallagher PG, Mohandas N. Hereditary spherocytosis. Lancet. 2008;372(9647):1411–1426. doi: 10.1016/S0140-6736(08)61588-3
- Wang C, Cui Y, Li Y, et al. A systematic review of hereditary spherocytosis reported in Chinese biomedical journals from 1978 to 2013 and estimation of the prevalence of the disease using a disease model. Intractable Rare Dis Res. 2015;4(2):76–81. doi: 10.5582/irdr.2015.01002
- Narla J, Mohandas N. Red cell membrane disorders. Int J Lab Hematol. 2017;39(Suppl.1):47–52. doi: 10.1111/ijlh.12657
- Agarwal AM. Ankyrin mutations in hereditary spherocytosis. Acta Haematol. 2019;141(2):63–64. doi: 10.1159/000495339
- Christensen RD, Yaish HM, Gallagher PG. A pediatrician’s practical guide to diagnosing and treating hereditary spherocytosis in neonates. Pediatrics. 2015;135(6):1107–1114. doi: 10.1542/peds.2014-3516
- He BJ, Liao L, Deng ZF, et al. Molecular genetic mechanisms of hereditary spherocytosis: current perspectives. Acta Haematol. 2018;139(1):60–66. doi: 10.1159/000486229
- Gallagher PG. Hematologically important mutations: ankyrin variants in hereditary spherocytosis. Blood Cells Mol Dis. 2005;35(3):345–347. doi: 10.1016/j.bcmd.2005.08.008
- Lambert S, Yu H, Prchal JT, et al. cDNA sequence for human erythrocyte ankyrin. Proc Natl Acad Sci USA. 1990;87(5):1730–1734. doi: 10.1073/pnas.87.5.1730
- Salas PC, Rosales JML, Milla CP, et al. A novel mutation in the beta-spectrin gene causes the activation of a cryptic 5′-splice site and the creation of a de novo 3′-splice site. Hum Genome Var. 2015;2:15029. doi: 10.1038/hgv.2015.29
- Bianchi P, Fermo E, Vercellati C, et al. Diagnostic power of laboratory tests for hereditary spherocytosis: a comparison study in 150 patients grouped according to molecular and clinical characteristics. Haematologica. 2012;97(4):516–523. doi: 10.3324/haematol.2011.052845
- Eber S, Lux SE. Hereditary spherocytosis – defects in proteins that connect the membrane skeleton to the lipid bilayer. Semin Hematol. 2004;41(2):118–141. doi: 10.1053/j.seminhematol.2004.01.002
- Morle L, Bozon M, Alloisio N, et al. Ankyrin Bugey: a de novo deletional frameshift variant in exon 6 of the ankyrin gene associated with spherocytosis. Am J Hematol. 1997;54(3):242–248. doi: 10.1002/(SICI)1096-8652(199703)54:3<242::AID-AJH11>3.0.CO;2-F
- Miya K, Shimojima K, Sugawara M, et al. A de novo interstitial deletion of 8p11.2 including ANK1 identified in a patient with spherocytosis, psychomotor developmental delay, and distinctive facial features. Gene. 2012;506(1):146–149. doi: 10.1016/j.gene.2012.06.086
- Iolascon A, Avvisati RA. Genotype/phenotype correlation in hereditary spherocytosis. Haematologica. 2008;93(9):1283–1288. doi: 10.3324/haematol.13344
- Bogusławska DM, Heger E, Sikorski AF. Molecular mechanism of hereditary spherocytosis. Pol Merkur Lekarski. 2006;20(115):112–116.
- Luo Y, Li Z, Huang L, et al. Spectrum of ankyrin mutations in hereditary spherocytosis: a case report and review of the literature. Acta Haemotol. 2018;140:77–86. doi: 10.1159/000492024
- Delaunay J. The molecular basis of hereditary red cell membrane disorders. Blood Rev. 2007;21(1):1–20. doi: 10.1016/j.blre.2006.03.005
- Shin S, Jang W, Kim M, et al. Targeted next-generation sequencing identifies a novel nonsense mutation in SPTB for hereditary spherocytosis. Medicine. 2018;97:S3–S8. doi: 10.1097/MD.0000000000009218
- Bolton-Maggs PH, Langer JC, Iolascon A, et al. Guidelines for the diagnosis and management of hereditary spherocytosis – 2011 update. Br J Haematol. 2012;156(1):37–49. doi: 10.1111/j.1365-2141.2011.08921.x
- Manciu S, Matei E, Trandafir B. Hereditary spherocytosis - diagnosis, Surgical treatment and outcomes. a literature review. Chirurqia(Bucur). 2017;112(2):110–116.