
ABSTRACT
Objectives: Acquired hemophilia A (AHA) is a rare disease resulting from autoantibodies against coagulation factor VIII that leads to spontaneous bleeding. This study reports the clinical characteristics and treatment outcomes of a relatively sizable cohort of patients with AHA.
Methods: We retrospectively analyzed the characteristics and outcomes of 42 patients with AHA diagnosed in our center from January 2014 through December 2018.
Results: The FVIII activity (FVIII: C) was significantly suppressed (median 1.5%; interquartile range [IQR]: 0.9–3.5) by FVIII inhibitor (median 8 BU/mL; IQR: 4.0–16.0). Bypassing agents, PCC or FVIIa, were used in 14 patients for bleeding control without any adverse reaction; and most patients (90.5%, 38/42) were placed on immunosuppressive regimen, corticosteroid alone or in combination with cyclophosphamide. Patients treated with corticosteroids alone had a lower median inhibitor titer (8 BU/mL) than those treated with combination corticosteroids of cyclophosphamide (16 BU/mL) (p < 0.001). 97.4% (37/38) patients achieved complete remission (CR) after immunosuppression therapy, and the median time to CR in patients treated with corticosteroids alone was shorter than those with combination corticosteroids of cyclophosphamide (median 40 days; IQR: 31–65 vs. 51 days; IQR: 38–83, p = 0.301). 10 (26.3%) patients relapsed thereafter and were placed on combined corticosteroid and cyclophosphamide treatment, which yielded second remission in 8 patients (80%). Two patients died, one from uncontrolled post-surgical retroperitoneal hemorrhage and one from sepsis complicating corticosteroid therapy.
Conclusion: The corticosteroid achieves a satisfactory outcome, particularly with low inhibitors titers; and combination of cyclophosphamide will facilitate remission in sever patients with high titers of inhibitors.
Introduction
Acquired hemophilia A (AHA) is a rare autoimmune disease with an estimated incidence of approximately 1.3–1.5 per million population per year, which is caused by autoantibodies against coagulation FVIII and characterized by spontaneous hemorrhage without family or personal history of bleeding [Citation1,Citation2]. AHA-related hemorrhages are associated with high morbidity and mortality [Citation3–5], and risk of bleeding persists in the presence of inhibitors. An underlying cause is detected in approximately 50% of cases including autoimmune diseases, malignancy, infections, postpartum complication and others [Citation6–10]. The FVIII: C and the inhibitor titer could not be measured in many hospitals due to the limited facility and resources, particularly in developing countries. Under recognition due to the rareness of the disease and lack of expertise in management often delayed the diagnosis and unsatisfactory outcome of treatment [Citation11]. The main goals of AHA therapy are to control acute bleeding and eliminate inhibitors [Citation12]. Large randomized clinical trials are considered unfeasible due to the rarity of AHA. To date, the largest retrospective study looking at AHA from the European Acquired Hemophilia Registry (EACH2), including 501 patients at 117 centers over 5 years revealed that remission was achieved faster with the combination of corticosteroids and cyclophosphamide than with corticosteroids alone, with a similar overall mortality rate [Citation13]. We have conducted a retrospective study summarizing the clinical characteristics and treatment outcomes of a relatively sizable cohort of Chinese patients with AHA.
Materials and methods
Patients
Complete medical records and laboratory data of 42 AHA patients, including 25 males and 17 females (age range: 10–79 years), who were referred consecutively to our hospital for hematology consultancy between January 2014 and December 2018 and followed for a median of 16.8 months, were retrieved and reviewed. The baseline characteristics, coagulation parameters, FVIII: C and FVIII inhibitor levels of the study population are described in . The age distribution is demonstrated in . The co-morbidities were classified into the following categories: spontaneous, surgical, traumatic injury, autoimmune disease, postpartum and others. Acute episodes were defined as new-onset of bleeding which required intervention and treatment to control. Severe bleeding referred to bleeding from visceral organs, the retroperitoneal compartment, intracranial hemorrhage or other life-threatening conditions. Control of acute bleeding was defined as hemostasis, no increase in hematoma size and the absence of new bleeding signs. The study was approved by the Research Ethics Committee and performed in accordance with the principles of the declaration of Helsinki.
Figure 1. Age distribution at diagnosis of 42 patients with AHA The age distribution showed inhibitors are typically biphasic with a larger peak in patients aged 20–40 years and another peak in patients aged 60–80 years.
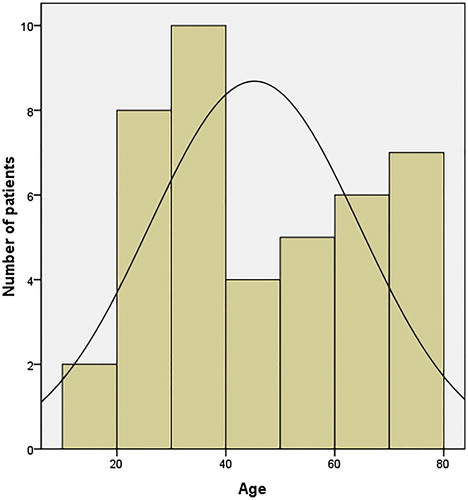
Table 1. Baseline characteristics of the 42 patients.
Inhibitor detection and diagnosis establishment
The coagulation status of patients was evaluated with global coagulation function assays, including activated partial thromboplastin time (APTT, Actin®, Siemens Healthcare Diagnostics, Marburg, Germany) and prothrombin time (Thromborel® S, Siemens Healthcare Diagnostics) using CS-5100 automatic coagulation analyzer (Sysmex Medical Co., Ltd., Kobe, Japan). The FVIII clotting activity was measured by one-stage APTT assay, using HemosIL® FVIII deficient plasma (IL Company, Bedford, MA, USA) on ACL TOP 700 coagulation analyzer (Werfen Group, Bedford, MA, USA). The presence of inhibitors was screened by APTT assay in either time-dependent or -independent fashion. The mixtures of patient plasma and normal pooled plasma (1:1) were either subjected to APTT assay immediately upon sample withdrawn or tested after 2 hours incubation at 37°C. Lupus anticoagulants (LAC) and acquired von Willebrand syndrome (vWF) were ruled out using HemosIL® dilute Russell viper venom time (dRVVT) screen/confirm, HemosIL® vWF:Ag and HemosIL® vWF Activity (IL Company) on ACL TOP 700 coagulation analyzer. The titer of the inhibitor was determined following standard Bethesda protocol [Citation14], AHA diagnosis was established with clinical presentations and laboratory findings, including reduced FVIII:C and the presence of FVIII inhibitor.
Hemostatic and immunosuppressive treatments
Due to the unavailability of activated prothrombin complex concentrate (aPCC) (FEIBA; FVIII inhibitor bypassing activity) in China, the prothrombin complex concentrate (PCC) (Taibang, Shandong Taibang Biological Products Co., Ltd., China; or Kangshuning, Hualan Biological Engineering Co., Ltd., China) at a dose of 25–50 U/kg/day [Citation11] and recombinant factor VIIa (rFVIIa, NovoSeven, Novo Nordisk A/S, Bagsvaerd, Denmark) were used for acute bleeding control at a dose of 90 µg/kg every 2–3 hours until hemostasis achieved. Human FVIII concentrate (Haemate P; Behringwerke, Marburg, Germany) were administered to patients with low-titer inhibitors.
Patients with lower inhibitor titer (median 8BU, IQR: 3–8) were placed on corticosteroid therapy alone. The initial dose of prednisone was 1 mg/kg as treatment guidance [Citation15] recommended. Patients who did not have inhibitor titer decreased after 2–3 weeks of treatment were switched to the regimen with combined prednisone and cyclophosphamide (200 mg/week). For patients with higher inhibitor titers (median 16BU, IQR: 16–32), combined treatment of 1 mg/kg prednisone and cyclophosphamide 200 mg/week were used. The antiCD20 antibody rituximab was used as the second-line agent for refractory or unsuitable cases for cytotoxic therapy. The complete remission (CR) was defined as resolution of hemorrhagic signs, recovery of FVIII activity (FVIII: C > 60%) and negative results from Bethesda assay. The corticosteroid was tapered gradually with dose halved every 2–3 weeks after the inhibitor was negative, and the inhibitor assay was performed before each dosing adjustment. If there was no relapse of inhibitors, the patients would be placed on 0.1 mg/kg corticosteroid for up to 4–5 weeks at the end of the treatment regimen. The FVIII: C and inhibitor would be followed for another 2–3 months after completion of the immunosuppressive therapy. Patients who had inhibitor titer increased with/without FVIII:C decrease were considered to have AHA relapsed and would be placed on combined corticosteroid and cyclophosphamide therapy no matter what the initial treatment regimen they had in first place.
Statistical analysis
Continuous variables were expressed as the mean value ± standard deviation (SD) or medians with IQRs according to the normality of data distribution. Groups were compared using Mann–Whitney U test for nonparametric variables. Survival curves were plotted using the Kaplan–Meier method and log-rank test. P < .05 was considered to indicate a statistically significant difference. All calculations were made with the SPSS 16.0 statistical package.
Results
Patient characteristics
All the 42 patients had prolonged APTT values (mean ± SD, 81.8 ± 18.4 seconds), decreased FVIII: C (median 1.5%; IQR: 0.9–3.5) and FVIII inhibitor of various titers (median 8.0 BU/mL; IQR: 4.0–16.0). The ecchymosis was the most frequent bleeding presentation (76.2%), followed by muscular hematoma (28.8%); urologic (23.8%); hemarthrosis (19.1%); gastrointestinal (9.5%) and retroperitoneal (4.8%). Eight patients (19.0%) had life-threatening bleeding upon diagnosis with two patients experience hemorrhagic shock due to postpartum bleeding and uncontrolled post-surgical retroperitoneal hemorrhaging. There was no significant difference in FVIII inhibitor titers between patients with severe hemorrhage and patients with non-severe bleeding presentations (median 12 BU; IQR: 8–16 vs. median 8 BU; IQR: 4–16, p = .459).
Treatment
Ninety-one (45.2%) of 42 patients required haemostatic support. Hemostatic therapy with bypassing agents was started in 14 (33.3%) of 42 patients to stop bleeding at presentation. Thirteen patients were given PCC alone with low dose of 25–50 U/kg/d. The median time to achieve hemostasis was 4 days (IQR: 3–6). The rFVIIa was given to a patient with postpartum bleeding which could not be controlled by the extended use of PCC for 10 days. Overall, 13 (92.9%) of 14 patients was controlled successfully with a bypassing agent for the bleeding episodes. One patient was died on day 10 due to uncontrolled post-surgical retroperitoneal hemorrhaging. Recombinant human FVIII was successfully used to treat bleeding in five patients with low-titer inhibitors.
Thirty-eight (90.5%) of 42 patients received immunosuppressive therapy. Corticosteroids alone in 21 patients (mean inhibitors titer: 8 BU/mL). Corticosteroids combined with cyclophosphamide in 10 patients (mean inhibitors titer: 16 BU/mL). Five patients (mean inhibitors titer: 16 BU/mL) were initially treated with corticosteroid but had cyclophosphamide added later to the treatment regimen. Two of them failed to have FVIII: C increased and inhibitor titer decreased after three weeks of corticosterone treatment. Two patients had AHA developed when they were on corticosterone therapy due to the presence of autoimmune disorders, paraneoplastic pemphigus and idiopathic thrombocytopenic purpura (ITP), respectively. One patient had FVIII inhibitor detected during the steroid management of reactive arthritis and acute generalized exanthematous pustulosis (AGEP) secondary to sepsis caused by Staphylococcus aureus, and cyclophosphamide was administered when the diagnosis of AHA was confirmed. Two patients had Sjögren’s syndrome and epidermolysis bullosa were not responsive to corticosteroid alone and inhibitor titer remained unchanged after the treatment. Rituximab was then added to treatment regimen, and the inhibitor titer decreased after 4 weeks of therapy (100 mg per week). The patients were then maintained with corticosterone alone ().
Table 2. Immunosuppression therapies and response of the 38 patients.
The 4 patients did not receive any standard immunosuppressive treatment. One patient with inhibitor titer of 16 BU/mL died in the day 10 of diagnosis before incipient of immunosuppression therapy due to uncontrolled post-surgical retroperitoneal hemorrhaging. Two male patients, aged 62 and 25 years, respectively, with low titers of inhibitors (<2) and one 71-year-old female with 8 BU/mL inhibitor and hematuria refused to take immunosuppressive treatment and were only managed with hemostatic therapy. The median time for the inhibitor to disappear for these patients was 77 days (IQR: 67–95).
Outcome of immunosuppression therapy
97.4% (37/38) patients achieved CR with immunosuppression therapy, the median time to CR in patients treated with corticosteroids alone, combined corticosterods and cyclophosphamide was 40 days (IQR: 31–65) and 51 days (IQR: 38–83) (). Patients who were unresponsive to corticosteroids therapy and had combined cyclophosphamide later in the treatment had much longer time to achieve CR (80 days, IQR: 53–97). One patient receiving corticosteroids alone treatment for 45 days died from sepsis before achieving a response.
Figure 2. Treatment outcome of corticosteroid alone and combination of corticosteroid and cyclophosphamide Kaplan–Meier plot showed that there was no difference between treatment groups in terms of time to achieve CR (Log-rank test, p = 0.344), although patients receiving combined corticosteroid and cyclophosphamide therapy having higher titers of FVIII inhibitors.
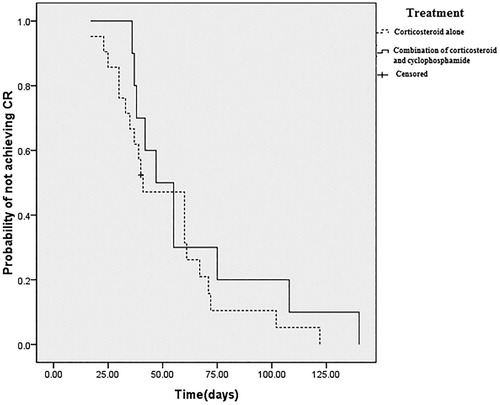
14.3% (3/21) of patients who treated only with corticosteroids became relapse, while 30.0% (3/10) of patients receiving combined corticosteroids and cyclophosphamide and 80.0% (4/5) of patients who had cyclophosphamide later added in treatment regimen had inhibitor titer increased again during follow up. Of total 10 relapsed patients, 80.0% (8/10) had inhibitors eliminated again with combined corticosteroid and cyclophosphamide treatment (). To the current, the overall CR rate was 92.1% (35/38).
Apart from one the death from sepsis on corticosteroids alone referred to above, there were few severe complications encountered during immunosuppressive treatment. Occasional neutropenia was observed in two patients and one patient had an episode of hemorrhagic cystitis.
Discussion
AHA is an autoimmune disorder from the adaptive immune system mediated by polyclonal IgG antibodies targeted at multiple antigenic sites presenting within the A2, C1 and C2 domains of FVIII [Citation16]. The AHA maybe predisposed with abnormality of immune system, as observed in the current study, an underlying medical condition, such as autoimmune diseases (Sjögren’s syndrome, ITP, paraneoplastic pemphigus, epidermolysis bullosa, etc.), infection, surgery, trauma, postpartum and drugs, all could lead to the development of FVIII inhibitors. Knoebl’s study on 501 AHA patients that age distribution in AHA patients are biphasic with more patients diagnosed in age range 20–40 years and 60–80 years with the median age of 73.9 years [Citation13]. Although the AHA patient cohort of the current study also has a wide age range from 10 to 79 years, the median age is much younger at 42 years. Whether the discrepancy could be attributed to the earlier presentation in a Chinese ethnicity or just poor case detection/referral for treatment in more elderly population demands further study to clarity. The bleeding presentations of AHA patients are different from those of congenital hemophilia [Citation17]. Similar to previous reports [Citation18,Citation19], subcutaneous bleeds were the most common bleeding site in patients reviewed in the current study. The severity of bleeding is not necessarily correlated to the titer inhibitors and residual FVIII activities, which might partly due to the complex type II kinetic of the inhibitor resulting in a rapid and non-linear inactivation of FVIII [Citation18].
An early recognition and diagnosis are critical to the management of AHA. Besides prompt control of bleeding, eradication of FVIII inhibitors is essential to the cure of AHA [Citation20]. FVIII replacement is not able to rescue bleeding in most patients with high titers of inhibitors and bypassing agents are the only effective option under such circumstances. The rFVIIa and aPCC are recommended for bleeding control of AHA [Citation21–23], particularly for life-threatening hemorrhages. Because of unavailability of aPCC in China and high cost of rFVIIa, PCC is instead used frequently in management of AHA patients at a relatively low dose of 25–50 U/kg/d. The PCC achieved hemostasis and ceased bleeding in patients in current study. However, repeated and extended treatment was required in most cases. The administering of bypassing agent was associated low incidence of thrombotic complications [Citation10,Citation24] and Knoebl reported thromboembolic events occurred in about 4% of cases treated with bypassing agents [Citation13]. Fortunately, no thrombotic events were recorded for the use of bypassing agent in patients’ cohort of current study.
Immunosuppression with corticosteroid is a primary therapy for inhibitor eradication. There are conflict reports on the benefit of combined use of corticosteroid with cyclophosphamide [Citation25]. The UKHCDO study showed no difference in inhibitor eradication or mortality between patients between corticosteroids alone and corticosteroids/ cyclophosphamide agents, and the combination of corticosteroids and cyclophosphamide did achieve CR more successfully and quicker than corticosteroids alone [Citation18]. It was suggested that whether to include cyclophosphamide in the treatment regimen should be based on patient characteristics such as age, general condition, and potential tolerance of therapy [Citation26]. The titer of FVIII inhibitor upon diagnosis may be another important factor in choice of treatment regimen. A French study showed that patients with inhibitor titers less than 20 BU/mL could achieve faster CR by treatment with corticosteroids alone and patients with FVIII < 1 IU/dL and/or inhibitor titers >20 BU/mL may need longer time to CR while combination of corticosteroids and immunosuppressant for inhibitor eradication [Citation27]. A German-Austrian study enrolled 102 prospectively patients showed partial remission(PR) was achieved at a median of 31 days, and patients with baseline FVIII ≥1 IU/dL achieved PR more often and quick than patients with <1 IU/dL. Further analysis demonstrated that low FVIII remained associated with a lower rate of PR, CR and decreased survival [Citation28]. We have placed patients with higher titers of FVIII inhibitors (medium 16 BU) and those respond poorly to corticosteroid alone therapy on combined regimen with both corticosteroid and cyclophosphamide. The threshold for addition of cyclophosphamide was a post hoc observation of the study. The CR rate and time to CR were similar in the corticosteroids alone and corticosteroids and cyclophosphamide combination group, although patients of latter group had higher titer of inhibitor. For patients who had reappeared inhibitors during tapering of corticosteroid therapy, addition of cyclophosphamide also helped to enable second CR in those patients. It seems that addition of cyclophosphamide will facilitate the remission in AHA patients, particularly those with high titer inhibitors.
Conclusion
AHA is a rare and heterogeneous disorder. Bleeding management with PCC may be an effective option where aPCC and rFVIIa are not available, particularly in developing countries. Immunosuppression with corticosteroid and cyclophosphamide can successfully eradicate FVIII inhibitors and achieve complete remission of AHA, particularly in patients with high titers of inhibitors. The well-designed prospective study is needed to provide a deeper insight into the demographics and therapeutic management of AHA in China.
Acknowledgments
The authors are thankful for the technique support of Guanqun Xu, BA, in coagulation function tests and Zhenzhen liu, MS for clinical data collection.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Saxena R, Mishra DK, Kashyap R, et al. Acquired haemophilia – a study of ten cases. Haemophilia. 2000;6:78–83. doi: 10.1046/j.1365-2516.2000.00372.x
- Tiede A, Worster A. Lessons from a systematic literature review of the effectiveness of recombinant factor VIIa in acquired haemophilia. Ann Hematol. 2018;97:1889–1901. doi: 10.1007/s00277-018-3372-z
- Collins P, Baudo F, Huth-Kuhne A, et al. Consensus recommendations for the diagnosis and treatment of acquired hemophilia A. BMC Res Notes. 2010;3:161. doi: 10.1186/1756-0500-3-161
- Zeitler H, Ulrich-Merzenich G, Goldmann G, et al. The relevance of the bleeding severity in the treatment of acquired haemophilia – an update of a single-centre experience with 67 patients. Haemophilia. 2010;16:95–101. doi: 10.1111/j.1365-2516.2008.01922.x
- Jayakar JP, O'Neill N, Yan M, et al. Retrospective review of acquired haemophilia A from the largest Canadian haemophilia treatment centre. Haemophilia. 2018;24:e383–e387. doi: 10.1111/hae.13598
- Kessler CM, Knobl P. Acquired haemophilia: an overview for clinical practice. Eur J Haematol. 2015;95(Suppl 81):36–44. doi: 10.1111/ejh.12689
- Kose M, Bakkaloglu OK, Amikishiyev S, et al. Acquired FVIII and FIX inhibitors after pregnancy: a case report. Acta Haematol. 2016;136:229–232. doi: 10.1159/000445706
- Rivera Cora NI, Irizarry Delgado F, Merle Ramirez SM, et al. Acquired hemophilia A in an advanced age patient of Hispanic origin: a case report. BMC Res Notes. 2017;10:438. doi: 10.1186/s13104-017-2767-6
- Takeyama M, Nogami K, Kajimoto T, et al. First report of real-time monitoring of coagulation function potential and IgG subtype of anti-FVIII autoantibodies in a child with acquired hemophilia A associated with streptococcal infection and amoxicillin. Int J Hematol. 2018;107:112–116. doi: 10.1007/s12185-017-2273-6
- Napolitano M, Siragusa S, Mancuso S, et al. Acquired haemophilia in cancer: a systematic and critical literature review. Haemophilia. 2018;24:43–56. doi: 10.1111/hae.13355
- Yang Y, Xue F, Shi H, et al. Acquired hemophilia A: retrospective analysis of 49 cases from a single Chinese hemophilia center. Clin Appl Thromb Hemost. 2015;21:35–40. doi: 10.1177/1076029613488937
- Charlebois J, Rivard GE, St-Louis J. Management of acquired hemophilia A: review of current evidence. Transfus Apher Sci. 2018;57:717–720. doi: 10.1016/j.transci.2018.10.011
- Knoebl P, Marco P, Baudo F, et al. Demographic and clinical data in acquired hemophilia A: results from the European Acquired Haemophilia Registry (EACH2). J Thromb Haemost. 2012;10:622–631. doi: 10.1111/j.1538-7836.2012.04654.x
- Tiede A, Werwitzke S, Scharf RE. Laboratory diagnosis of acquired hemophilia A: limitations, consequences, and challenges. Semin Thromb Hemost. 2014;40:803–811. doi: 10.1055/s-0034-1390004
- Kruse-Jarres R, Kempton CL, Baudo F, et al. Acquired hemophilia A: updated review of evidence and treatment guidance. Am J Hematol. 2017;92:695–705. doi: 10.1002/ajh.24777
- Kahle J, Orlowski A, Stichel D, et al. Frequency and epitope specificity of anti-factor VIII C1 domain antibodies in acquired and congenital hemophilia A. Blood. 2017;130:808–816. doi: 10.1182/blood-2016-11-751347
- Wang XF, Zhao YQ, Yang RC, et al. The prevalence of factor VIII inhibitors and genetic aspects of inhibitor development in Chinese patients with haemophilia A. Haemophilia. 2010;16:632–639.
- Collins PW, Hirsch S, Baglin TP, et al. Acquired hemophilia A in the United Kingdom: a 2-year national surveillance study by the United Kingdom Haemophilia Centre Doctors’ Organisation. Blood. 2007;109:1870–1877. doi: 10.1182/blood-2006-06-029850
- Kessler CM, Ma AD, Al-Mondhiry HA, et al. Assessment of acquired hemophilia patient demographics in the United States: the Hemostasis and Thrombosis Research Society Registry. Blood Coagul Fibrinolysis. 2016;27:761–769. doi: 10.1097/MBC.0000000000000582
- Knobl P. Prevention and management of bleeding episodes in patients with acquired hemophilia A. Drugs. 2018;78:1861–1872. doi: 10.1007/s40265-018-1027-y
- Ma AD, Kessler CM, Al-Mondhiry HA, et al. Use of recombinant activated factor VII for acute bleeding episodes in acquired hemophilia: final analysis from the Hemostasis and Thrombosis Research Society Registry acquired hemophilia study. Blood Coagul Fibrinolysis. 2016;27:753–760. doi: 10.1097/MBC.0000000000000471
- Zanon E, Pasca S, Siragusa S, et al. Low dose of aPCC after the initial treatment in acquired haemophilia A is useful to reduce bleeding relapses: data from the FAIR registry. Thromb Res. 2018;174:24–26. doi: 10.1016/j.thromres.2018.12.006
- Zanon E, Milan M, Gamba G, et al. Activated prothrombin complex concentrate (FEIBA(R)) for the treatment and prevention of bleeding in patients with acquired haemophilia: A sequential study. Thromb Res. 2015;136:1299–1302. doi: 10.1016/j.thromres.2015.10.032
- Arokszallasi A, Razso K, Ilonczai P, et al. A decade-long clinical experience on the prophylactic use of activated prothrombin complex concentrate in acquired haemophilia A: a case series from a tertiary care centre. Blood Coagul Fibrinolysis. 2018;29:282–287.
- Franchini M, Vaglio S, Marano G, et al. Acquired hemophilia A: a review of recent data and new therapeutic options. Hematology. 2017;22:514–520. doi: 10.1080/10245332.2017.1319115
- Elezovic I. Acquired haemophilia syndrome: pathophysiology and therapy. Srp Arh Celok Lek. 2010;138(Suppl 1):64–68. doi: 10.2298/SARH10S1064E
- Vautier M, de Boysson H, Creveuil C, et al. Influence of factor VIII level and its inhibitor titer on the therapeutic response to corticosteroids alone in the management of acquired hemophilia: A retrospective single-center study. Medicine (Baltimore. 2016;95:e5232. doi: 10.1097/MD.0000000000005232
- Tiede A, Klamroth R, Scharf RE, et al. Prognostic factors for remission of and survival in acquired hemophilia A (AHA): results from the GTH-AH 01/2010 study. Blood. 2015;125:1091–1097. doi: 10.1182/blood-2014-07-587089