
ABSTRACT
Objective: To summarize the abnormal location of FLT3 caused by different glycosylation status which further leads to the distinguishing signaling pathways and discuss targeting on FLT3 glycosylation by drugs reported in recent literatures.
Methods: We review FLT3 glycosylation in endoplasmic reticulum. The abnormal signal of mutant FLT3 with different glycosylation status is discussed. We also address potential FLT3 glycosylation-targeting strategies for the treatment.
Results: Inhibition of FLT3 mutant cells by drugs reported in recent literatures involves the influence of glycosylation of FLT3: 2-deoxy-D-glucose, Tunicamycin and Fluvastatin are reported to inhibit N-glycosylation of FLT3; Pim-1 inhibitors are proved to block the inhibition of Pim-1 on FLT3 Oglycosylation; HSP90 inhibitors and Tyrosine Kinase Inhibitors are shown to increase fully glycosylated form of FLT3.
Discussion: The FMS-like tyrosine kinase 3 (FLT3) gene expressed only in CD34+ progenitor cells in bone marrow is located on chromosome 13q12 encoding FLT3 protein. FLT3 is initially synthesized as a 110 KD protein, which glycosylated in the endoplasmic reticulum to a 130 KD immature protein rich in mannose, and further processed into a mature 160 KD protein in the Golgi apparatus, which could be transferred to the cell surface. Therapy targeting on FLT3 glycosylation is a promising direction for AML treatment.
Conclusions: The abnormal location of FLT3 caused by different glycosylation status leads to the distinguishing signaling pathways. Targeting on FLT3 glycosylation may provide a new perspective for therapeutic strategies.
Abbreviations: ABCG2: ATP-binding cassette transporter breast cancer resistance protein; ATF: activating transcription factor; AML: acute myeloid leukemia; CHOP: CCAAT-enhancer-binding protein homologous protein; 2-DG: 2-deoxy-D-glucose; EFS: event free survival; EPO: erythropoietin; EPOR: erythropoietin receptor; ERS: endoplasmic reticulum stress; FLT3: FMS-like tyrosine kinase 3; GPI: glycosylphosphatidylinositol; HSP: heat shock protein; ITD: internal tandem duplication; IRE1a: inositol-requiring enzyme 1 alpha; JNK: c-Jun N-terminal kinase; JMD: juxtamembrane domain; JAK: janus kinase; MAPK/ERK: mitogen activated protein kinase/extracellular signal-regulated protein kinase; OS: overall survival; PI3K/AKT: phosphatidylinositide 3-kinases/protein kinase B; PERK: RNA-activated protein kinase-like endoplasmic reticulum kinase; Pgp: P-glycoprotein; PTX3: human pentraxin-3; STAT: signal transducer and activator of transcriptions; TKD: tyrosine-kinase domain; TKI: tyrosine kinase inhibitor; TM: Tunicamycin; UPR: unfolded protein reaction
1. Introduction
Acute myeloid leukemia (AML) is a heterogeneous malignant clonal disorder that starts from bone marrow immature myeloid progenitor cells transformed by recurrent genetic alterations to induce leukemic cell proliferation and survival [Citation1]. These cells are capable of self-renewing and sustaining malignant populations as well as producing subclones [Citation2]. Approximately 40–45% of younger and 10–20% of older adults with acute myeloid leukemia (AML) could be cured with current standard chemotherapy [Citation3]. The basis of curative treatment is intensive combination chemotherapy comprising cytarabine and anthracycline (‘7 + 3’ regimen). Activated FLT3 mutation which is one of the most common mutant genes in AML occurs in about 40% of patients with acute myeloid leukemia (AML) with normal cytogenetics [Citation4,Citation5]. There are some ALL-T (pre-T) patients with mutated FLT3-ITD also, the prognosis of these patients is gloomy.
FMS-like tyrosine kinase 3 (FLT3) is a member of the tyrosine kinase receptor type III family and makes a great effect on the proliferation and differentiation of early hematopoietic progenitor cells [Citation6]. The FLT3 gene expressed only in CD34+ progenitor cells in bone marrow is located on chromosome 13q12 encoding FLT3 protein. When ligands bind to the extracellular domain of FLT3 receptor, the FLT3 receptor is dimerized and the tyrosine kinase domain is activated. A series of intracellular signaling molecules are activated through JAK/STAT, PI3K/AKT and MAPK/ERK signal transduction. The pathway regulates the growth and development of hematopoietic stem/progenitor cells [Citation7]. The ability of stem cells lacking FLT3 to reconstruct T cells and myelocytes was observed to decrease. Therefore, FLT3 may also play an important role in the development of pluripotent stem cells and B cells [Citation8].
Glycosylation is a modification occurring in post-translational proteins. The glycoproteins are involved in many key biological processes, including cell growth, differentiation and immune regulation [Citation9]. Altered glycosylation has been found in inflammatory diseases and many types of cancer [Citation10,Citation11]. Glycosylation modification of proteins with the participation of a series of enzymes refers to the process of covalent binding of sugar chains to specific amino acid residues in proteins. Glycosylation of proteins is involved in cell recognition by affecting the spatial structure, localization and stability of new peptide chains. Abnormal glycosylation modification is closely related to the avoidance of surveillance and tumorigenesis [Citation12,Citation13]. Endoplasmic reticulum generates the initiation of many types of protein glycosylation, for example N-glycosylation, O-mannosylation, and glycosylphosphatidylinositol (GPI) anchor addition [Citation13].
FLT3 is synthesized into 130 KD glycosylation-deficient or mannose-rich species, then folded in the endoplasmic reticulum and exported to the Golgi apparatus, where it is glycosylated to form a complex 150 KD glycosylation species and then transferred to the cell surface [Citation14,Citation15]. If FLT3 cannot be glycosylated correctly in Golgi, FLT3 protein will accumulate in the endoplasmic reticulum, causing cells to respond to the unfolded proteins, thus promoting cell apoptosis [Citation16] or triggering signal pathway different from glycosylation species.
2. Structure and mutant type of FLT3
The FLT3 receptor consists of five different parts: an extracellular domain, a transmembrane region, a cytoplasmic juxtamembrane domain JMD, and 2 cytoplasmic tyrosine kinase domains TKDs [Citation17–19]. The extracellular domain consists of 5 immunoglobulin-like domains and 2 TKDs are interrupted by a short kinase insert. The JMD composes of 3 distinct parts: the JM-binding motif (JM-B), the switch motif (JM-S), and the linker/zipper peptide segment (JM-Z) [Citation20]. JM-B is related to the conformation of activated/inactivated and stably inactivated kinases; JM-S consists of two phosphorylated sites containing STAT5 binding motifs; JM-Z can rotate substantially around its attachment sites [Citation20] ().
Figure 1. Structure of FLT3. ED, extracellular domain; TD, transmembrane domain; JMD, juxtamembrane domain; TKD1, tyrosine kinase domain-1; KI, kinase insert; TKD2, tyrosine kinase domain-2.
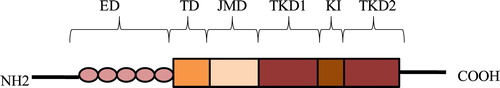
Activated FLT3 mutation which is one of the most common mutant genes in AML occurs in about 40% of patients with acute myeloid leukemia (AML) with normal cytogenetics [Citation4,Citation5]. There are three major groups of activation mutations: FLT3-internal tandem duplications (FLT3-ITDs) in the juxtamembrane (JM) domain in about 20% to 25% of patients [Citation21–23], FLT3 point mutations in the tyrosine-kinase domain (FLT3-TKD) in about 7% to 10% of patients [Citation24,Citation25] and point mutations in the juxtamembrane domain (JMD) and in the extracellular domain in less than 2% of patients [Citation26,Citation27].
The internal tandem duplication within the juxtamembrane domain of the receptor (JMD-ITD) was the first mutation in FLT3 and no other ITD mutations were found for many years [Citation21]. It is reported that the sites of internal tandem replication (ITD) in FLT3 juxtamembrane domain (JM) of AML patients occur in exon 11 and 12. FLT3-ITD is usually concentrated in the tyrosine-rich juxtamembrane region which encoded by codon 589–599. Several replicated nucleotides are inserted sequentially to prolong the JM region [Citation21]. Recent studies show that ITDs appear not only in the juxtamembrane region, but also in the kinase domain [Citation28]. About 30% of ITDs are located within the tyrosine kinase domain (TKD) according to the reports [Citation28]. It means replicated nucleotides embed not only into the JM region but also into the tyrosine kinase domain.
In addition to the common ITD mutation, the FLT3 gene mutation also has a point mutation involving the tyrosine kinase domain (TKD), which is found in 7% to 10% AML patients. The most common mutation was 835 aspartic acid (ASP835) residues [Citation25,Citation29]. Recently, two new point mutations are found in AML. One was the deletion of 836 isoleucine, the other was that 836 isoleucine was replaced by methionine and arginine [Citation29,Citation30]. These two new point mutations, like D835, are located in the activation loop of the second tyrosine kinase, which can lead to phosphorylation of the receptor itself in the absence of ligands and prevent apoptosis of AML cells by activating STAT5 [Citation30]. Other mutations such as F691, N676 usually occurred after tyrosine kinase inhibitors (TKIs) are used. As for other point mutations which located in the juxtamembrane domain (JMD) as well as in the extracellular domain appear with a very low frequency ().
Table 1. Mutant type of FLT3.
3. Prognosis and current treatments of mutant FLT3 in patients
All mutations can lead to dimerization and continuous autophosphorylation of FLT3 without ligand binding which leads to infinite cell proliferation. FLT3-ITDs in the juxtamembrane region is thought to destroy the self-suppressing conformation of FLT3 receptor, thereby activating downstream pathways continuously, including MAPK/ERK, STAT5 and PI3K [Citation31]. In addition, high expression of the wild type FLT3 usually self-activated in AML patients to induce abnormal activation of downstream signaling pathways that promote the proliferation of leukemia cells. The abnormal activation of signal pathways is closely related to prognosis.
The dynamic detection of FLT3 expression level in AML patients can be used as an indicator to judge the prognosis of AML and to monitor the small residual focus [Citation23,Citation32]. The AML patients with mutant FLT3-ITD have the characteristics of high leukocyte, high mortality rate, easy recurrence after chemotherapy and hematopoietic stem cell transplantation and short survival time in comparison to those with wild-type FLT3. Patients with ITD mutation in TKD (TKD-ITD) have worse survival prognosis than those in JMD, but the reason is not clear [Citation33]. But Marhäll et al. [Citation34] report that TKD-ITD shows similar carcinogenic potential to the JMD-ITD but a higher carcinogenic potential than the D835Y point mutation. The number of D835 point mutations in relapsed patients is relatively low, proving that FLT3 point mutations may not related to the development of the relapse [Citation35]. There is no significant correlation between the FLT3-TKD point mutation and the presence of hyper-leukocyte in the patients, but the patients’ disease-free survival is also shorter, and the signal transduction pathway is different from the FLT3-ITD mutation. Meanwhile, wild type FLT3 is also highly expressed in some AML patients of poor prognosis [Citation36].
Besides disease relapse is a common event even after allo-HSCT in patients with FLT3 – ITD mutant AML. Historically, patients with FLT – ITD AML have a higher rate of relapse after transplantation compared to patients with FLT3 WT (30% vs. 16%, respectively) [Citation37].
Aberrantly activated FLT3-kinase is considered to represent an attractive therapeutic target in AML. Several tyrosine kinase inhibitors (TKIs) are in development as targeted therapy for FLT3 mutant AML [Citation38]. Sorafenib (first generation of TKIs) has been shown to improve EFS, but not OS in younger adults with AML; Midostaurin (first generation of TKIs) improves OS with a HR of 0.78 [Citation39]. Crenolanib(second generation of TKIs) in combination with chemotherapy resulted in CR/CRi rates of 96% among FLT3 mutant subjects, Gilteritinib (second generation of TKIs) also produced CR/CRi rates of 89% [Citation39]. Though adding FLT3 inhibitors to standard frontline chemotherapy in FLT3 mutant AML patients results in a survival benefit [Citation37], unfortunately, FLT3-ITD TKIs only have a relatively modest and transient effect, indicating that TKIs do not completely eradicate LSCs. TKI resistance can occur as a result of mutations in the kinase domain of FLT3 [Citation38]. The treatment needs to be improved urgently.
4. FLT3 glycosylation in endoplasmic reticulum
The major modifications of proteins entering the endoplasmic reticulum include glycosylation, hydroxylation, acylation and disulfide bond formation. More than 50% of human proteins are glycosylated modified proteins [Citation40]. Glycosylation modification has greatly changed the physical and chemical properties of proteins: increased molecular weight and solubility. At the same time, it affects the function of proteins, such as glycosylation plays an important role in protein folding, transport and location [Citation41,Citation42]. According to the connection modes, it can be divided into four types: N-glycosylation, O-glycosylation, C-glycosylation and glycosylphosphatidylinositol (GPI) anchored connection [Citation43,Citation44]. FLT3 has been reported to be N-glycosylated in the endoplasmic reticulum [Citation45–47], which may be due to the difficulty in O-glycosylation research. FLT3 is initially synthesized as a 110 KD protein, which is glycosylated in the endoplasmic reticulum to a 130 KD immature protein rich in mannose, and further processed into a mature 160 KD protein in the Golgi apparatus, which could be transferred to the cell surface [Citation14]. Once on the surface, FLT3 binding to FL can lead to receptor dimerization, autophosphorylation and activation [Citation48]. FL combined with FLT3 can activate many downstream pathways, including PI3K/AKT, RAS/MAPK and JAK/STAT5 [Citation49–53]. Compared with wild type FLT3, FLT3-ITD is mainly expressed in the form of immature and insufficient glycosylation [Citation45]. The localization of wild type FLT3 and mutant FLT3 is different in cells.
5. Abnormal signal of mutant FLT3 with different glycosylation status
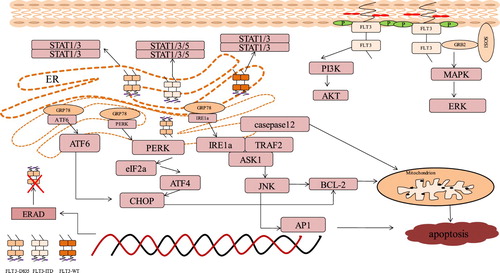
Wild-type FLT3 receptor proteins are mainly localized on the cell surface. Conversely, FLT3-ITD receptor proteins aggregate in the perinuclear region and are not found in the plasma membrane [Citation54]. It is reported that FLT3-ITD receptor protein is not confined to the endoplasmic reticulum or Golgi apparatus, but exists in an unknown compartment [Citation54]. But Schmidt-Arras et al. [Citation14] report that mutant FLT3-ITD protein is located at the endoplasmic reticulum and the Golgi apparatus. The differential location of FLT3 is related to different signal quality and endoplasmic reticulum interception [Citation15]. Mutant FLT3-ITD shows changes in signal quality, for FLT3-ITD located on cell surface in a small part and largely in endoplasmic reticulum, it maintains the strong constitutive activation of ERK1/2, AKT, STAT5, and to a lesser extent, STAT3 and STAT1. However, FLT3-ITD endoplasmic reticulum anchoring reduces the constitutive ERK1/2 and AKT activation [Citation15]. Endoplasmic reticulum anchoring FLT3-ITD produces aberrant signals like activation of STAT signaling pathways but have no ability to upregulate other signaling pathways such as ERK and AKT [Citation15,Citation55]. Consistent with FLT3-ITD, the endoplasmic reticulum retention of wild type FLT3 or D835Y caused activation of STAT1 and STAT3, but the constitutive signals of ERK1/2 and AKT are suppressed because of loss of ligand responsiveness in terms of autophosphorylation and concomitant activation. Endoplasmic reticulum-anchored FLT3-ITD still activates STAT5 compared to FLT3-WT and FLT3-D835Y [Citation56–60]. The predominance of STAT5 activation for transformation by FLT3-ITD has been indicated [Citation61,Citation62]. FLT3-D835Y and FLT3-ITD mutated receptors drives ERK and PI3K/AKT activation in a small part from the cell surface compared to high expression of wide type FLT3, whereas STAT3 and STAT5 signaling are induced predominantly from the endoplasmic reticulum [Citation55]. Mutant FLT3 activates the unfolded protein response (UPR). Correct protein folding and location depend on effective glycosylation. Without glycosylation, cells may respond to the presence of unfolded proteins, thus promoting cell apoptosis [Citation16]. The upstream signaling molecules are mainly endoplasmic reticulum proteins that bind to the molecular chaperone GRP78: PERK, ATF6 and IRE1a. UPR is activated in many cancer cells, but the mechanism of their role in cancer progression remains unclear.
Zhang et al. [Citation38] believe that oncoproteins cause UPR, while UPR causes HSP90 activation to stabilize oncoproteins and form positive feedback to cause cancer progression ().
Figure 2. The abnormal pathway triggered by mutant FLT3 in different glycosylation status. FLT3-ITD on cell surface in form of 160KD maintains the strong constitutive activation of ERK and AKT and endoplasmic reticulum anchoring FLT3-ITD activates STAT5, STAT3 and STAT1 to a lesser extent. Wild type FLT3 or D835Y in endoplasmic reticulum causes activation of STAT1 and STAT3, but the constitutive signals of ERK and AKT are suppressed compared to their form on cell surface. Mutant FLT3 activates the signaling molecules of the unfolded protein response (UPR): PERK, ATF6 and IRE1a to further trigger the downstream targets to induce apoptosis or ERAD system to degrade FLT3 protein.
6. Therapy targets on FLT3 glycosylation
Target on FLT3 glycosylation may provide a new perspective for therapeutic strategies ().
Table 2. Therapy targets on FLT3 glycosylation.
6.1. 2-deoxy-D-glucose
2-deoxy-D-glucose (2-DG) is known as one of sugar analogues to block N-linked glycosylation synthesis which plays an important role in inhibiting glucose metabolism [Citation63]. It is reported that hypoxic cancer cells are more dependent on glycolysis to provide enough ATP which can be blocked by 2-DG because of its similar structure to glucose; In the same manner, 2-DG can interfere with N-bond glycosylation because of its similar structure to mannose. 2-DG inhibits lipid-linked oligosaccharides and prevents mannose-type proteins from glycosylation.
Cell surface expression and signal transduction of FTL3-ITD can be both affected by 2-DG [Citation45]. During N-bond glycosylation, 2-DG mimics mannose by gradually adding it to lipid-linked oligosaccharide chains. In addition, high doses of 2-DG, rather than low glucose, hinders the O-glycosylation of cytosolic proteins, inhibiting tumor cells growth or promoting tumor cells death [Citation64]. The anticancer activity of 2-DG in solid tumors has also been confirmed [Citation65]. Normal folding of proteins is blocked by inhibiting N-bond glycosylation, which activates unfolded protein reaction (UPR) and retains these proteins in the endoplasmic reticulum [Citation66,Citation67]. 2-DG can inhibit N-bond glycosylation to affect protein glycosylation, resulting in the accumulation of misfolded proteins and further activating endoplasmic reticulum stress (ERS) to induce cells to apoptosis [Citation68–71].
6.2. Tunicamycin
Tunicamycin(TM) is a bacterial antibiotic that significantly inhibits the transfer of active sugars to dolichol phosphate, which is an important step in the N-glycosylation of protein at endoplasmic reticulum [Citation46,Citation47]. TM inhibits the synthesis of N-linked oligosaccharides in many cells [Citation72]. The expression of membrane receptors that show carcinogenicity can be decreased by TM [Citation73]. Downstream signaling pathways were interrupted by N-linked glycosylation suggesting that TM may be an alternative treatment to reduce carcinogenic signals and drug resistance [Citation74]. TM exhibited cytotoxic effects and enhance the susceptibility of therapy on different cell lines including human head and neck carcinoma and lung cancer cells through glycosylation inhibition [Citation75]. In lung cancer cells, deglycosylation of human pentraxin-3 (PTX3) protein by TM enhanced the sensitivity to therapy via AKT/NF-κB signaling pathway. TM inhibits glycosylation of plasma membrane receptors, resulting in impaired transport of these receptors to the cell surface [Citation76,Citation77].
In FLT3-ITD-harboring cells, ERS is triggered through activation of protein kinase RNA-like endoplasmic reticulum kinase (PERK) and CCAAT-enhancer-binding protein homologous protein (CHOP). TM arrested underglycosylated FLT3-ITD in an endoplasmic reticulum-bound form to activated STAT5 [Citation78]. But mere retention of FLT3 in the endoplasmic reticulum is not sufficient to activate signaling and mutation of FLT3 is necessary for initiation of downstream STAT5 signaling from the endoplasmic reticulum [Citation77]. In FLT3-ITD mutant cells, TM has been confirmed to arrest under-glycosylated FLT3-ITD in the endoplasmic reticulum and to promote STAT5 activation [Citation55,Citation78].
6.3. Fluvastatin
Statins act by blocking 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCoA reductase) [Citation79] which play an important role in the mevalonate pathway that generates dolichol related to N-linked glycosylation [Citation80]. Statins could inhibit proliferation of cancer cells in vitro and in vivo [Citation81–84]. Mevastatin can increase the sensitivity of patients with primary AML to standard therapy [Citation81]; Simvastatin increases the toxicity of chemotherapeutic drugs to colorectal cancer cells [Citation82]; Lovastatin can inhibit the expression of erythropoietin receptor by double inhibition of glycation [Citation85]. Erythropoietin (EPO) is essential for erythroid progenitor cells to survive by interacting with cell surface receptors. Recent literatures have shown that erythropoietin receptor (EPOR) is expressed in many cancer cells. Erythropoietin (EPO) that a member of the class I cytokine family is synthesized into 62 kDa precursors and glycosylated into 64 kDa proteins. The mature EpoR has 66 kDa molecular weight and complex Golgi-processed glycosylation mode [Citation86]. The mature EpoR in fully glycosylated state can be blocked by Lovastatin to prevent surface expression of EpoR [Citation85]. In addition, insulin and insulin-like growth factor (IGF) signaling occurs when intracellular receptor is under proper N-glycosylation of polyphenols. Especially at that time, statins significantly reduced the proliferation induced by IGF and insulin. Statins treatment resulted in the decreased expression of receptor membrane receptor, suggesting the destruction of glycation-dependent division [Citation87].
Similar with the EpoR, statins reduce the expression of mature and immature FLT3 in FLT3-positive cells because they inhibit the formation of dolichol. Statins can reduce the activity of mutant FLT3 kinases by blocking complex glycosylation of receptors, which in turn promotes the change of localization and signal transduction and leads to apoptosis [Citation16].
6.4. Pim-1 inhibitors
The oncogenic serine/threonine kinase proviral integration site for Moloney murine leukemia virus 1 (Pim 1) can regulate cytokine signaling in various cancers including myeloma, leukemia, prostate and breast cancers [Citation88]. In breast cancer, Pim1 is reported to activate the ATP-binding cassette transporter breast cancer resistance protein (ABCG2). Pim-1 also regulates the ABC transporter P-glycoprotein (Pgp). As a 150 kDa species, Pgp is exported from endoplasmic reticulum, which is glycosylated to 170kDa Pgp and transported to the cell surface. Pim-1 protects 150 kDa Pgp from proteolytic enzymes and proteasome degradation, resulting in glycosylation of Pgp and cell surface translocation [Citation89].
In AML, Pim-1 inhibition decreased the expression and half-life of 130 kDa FLT3 and increased 150 kDa FLT3 expression with abrogation by inhibition of glycosylation. FLT3 contains a common phosphorylation site of Pim-1 substrate [Citation90]. Serines undergo competitive O-linked glycosylation [Citation91], implying that mutation of serine at 935 of FLT3-ITD may largely inhibits its complex glycosylation by Pim-1, thereby retaining FLT3-ITD as a 130 KD form. Pim-1 may stabilize FLT3 in a 130KD form by inhibiting serine O-glycosylation, which can be destroyed by Pim-1 inhibitors [Citation92].
6.5. HSP90 inhibitors
Increased protein synthesis rate induced by carcinogenic signals often leads to protein toxicity stress, and HSP90 family proteins are essential to alleviate this stress and promote the survival of cancer cells. UPR restores cell homeostasis by degrading misfolded proteins and inducing the synthesis of chaperone proteins HSP90 [Citation93].
Wild-type FLT3 does not bind to HSP90 even under FL stimulation. FLT3-ITD has an abnormally prolonged JM domain because of the insertion of repetitive sequences into the JM region. HSP90 combines with FLT3-ITD to stabilize its structure, which makes FLT3-ITD accumulate abnormally in the endoplasmic reticulum [Citation94]. FLT3-ITD activates UPR to increase the expression of HSP90 which in turn stabilizes FLT3-ITD+ proteins to protect acute myeloid leukemia cells from apoptosis [Citation38]. HSP90 protects oncoproteins from misfolding and degradation. Inhibitors of HSP90 family proteins can lead to instability of FLT3 protein to further activate toxic overload triggering apoptosis induced by unfolded protein reaction [Citation95]. Inhibiting the function of HSP90 may result in incorrect folding or rapid degradation of related proteins [Citation96]. Failure of FLT3-ITD to translocate to the cell surface is accompanied by a pronounced glycosylation defect [Citation16]. HSP90 inhibitors result in a relative increase of FLT3 in localization of cell membrane [Citation38,Citation97].
6.6. Tyrosine kinase inhibitors
Targeted FLT3 seems to help minimally eradicate residual diseases, reduce recurrence rates and prolong event-free survival (EFS) and overall survival (OS) during maintenance treatment [Citation32]. Tyrosine Kinase Inhibitors (TKIs) are main therapy for FLT3 positive patients. Autophosphorylation of FLT3-ITD receptors may inhibit the physiological processes required for maturation and surface expression. FLT3-ITD is abnormally located in endoplasmic reticulum as an immature protein, but its molecular mechanism is not clear. FLT3-ITD maturation and cell surface expression were restored by treatment with FLT3 inhibitor TKIs [Citation98]. In antigen presentation of T cells, TKI therapy regulates post-translational glycosylation of MHC(major histocompatibility complex) molecules to modulate immune response [Citation99]. This helps to demonstrate that TKIs treatment increased the mature and complete glycosylation forms of FLT3 in FLT3-ITD positive cells [Citation32,Citation98].
It was found that the expression of FLT3 on the surface in TKI-resistant cells containing FLT3-ITD was decreased. FLT3-ITD with a new emerging point mutation showed a reduced FLT3 surface expression after TKI treatment and resistance to TKI therapy [Citation100].
7. Conclusion
The abnormal location of mutant FLT3 is caused by different glycosylation states, and finally activates the abnormal signaling pathway. FLT3 is an important gene to promote AML progression associated with poor prognosis in AML, but the mechanism of FLT3 in leukemia cell proliferation is still unknown. The presence of a FLT3 mutation has a strong negative prognosis in AML due to an increased risk of relapse after chemotherapy. Many tyrosine kinase inhibitors (TKIs) have been shown to inhibit FLT3/ITD phosphorylation. Wild-type FLT3 is often inhibited to a lesser extent by many FLT3 TKIs. However, some FLT3 TKI have very little activity against FLT3 kinase domain-activating mutations, especially D835Y, which makes leukemic cells resist [Citation16]. Some of them have already shown that FLT3 TKIs, such as sorafenib (first generation of TKI) or quizartinib (second generation of TKI) fail to induce long-lasting remissions [Citation37]. The prognosis of patients with mutant FLT3 is still poor. Thus, newer TKIs or a numerous class of drugs as a significant transformative component of malignancy are developed for the treatment of AML with mutant FLT3. Mutant FLT3 is known to have three main mutations, the most common of which is FLT3-ITD. Wild-type FLT3 mainly expresses on cell membrane, while mutant FLT3 accumulates in endoplasmic reticulum in the form of non-glycosylation. At present, it is generally believed that FLT3 can activate JAK/STAT, PI3K/AKT, MAPK/ERK three classical pathways. The change of signal pathway activated by mutant FLT3 is closely related to its abnormal location. FLT3 in endoplasmic reticulum activates JAK/STAT signaling pathway more than MAPK/ERK, PI3K/AKT. However, the unfolded protein reaction-related apoptosis and the self-degradation of FLT3 protein because of a large amount of FLT3 protein accumulated abnormally in endoplasmic reticulum is far less than the proliferation-promoting effect of JAK/STAT activated by endoplasmic reticulum-targeted FLT3. The mechanism of many recent drugs in FLT3 mutant-positive cells involves glycosylation, targeting on glycosylation of FLT3 may provide a new perspective for the treatment of patients with mutant FLT3.
Acknowledgements
The authors are very grateful to the members of the Leukemia Research Institute in Ren Ji Hospital.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Hospital MA, Green AS, Maciel TT, et al. FLT3 inhibitors: clinical potential in acute myeloid leukemia. Onco Targets Ther. 2017;10:607–615.
- Acheampong DO, Adokoh CK, Asante DB, et al. Immunotherapy for acute myeloid leukemia (AML): a potent alternative therapy. Biomed Pharmacother. 2018;97:225–232.
- Bose P, Vachhani P, Cortes JE. Treatment of relapsed/refractory acute myeloid leukemia. Curr Treat Options Oncol. 2017;18(3):17.
- Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002;100(5):1532–1542.
- Stirewalt DL, Radich JP. The role of FLT3 in haematopoietic malignancies. Nat Rev Cancer. 2003;3(9):650–665.
- Volpe G, Clarke M, Garcia P, et al. Regulation of the Flt3 gene in haematopoietic stem and early progenitor cells. PloS one. 2015;10(9):e0138257.
- Takahashi S. Downstream molecular pathways of FLT3 in the pathogenesis of acute myeloid leukemia: biology and therapeutic implications. J Hematol Oncol. 2011;4(13). doi:10.1186/1756-8722-4-13.
- Mackarehtschian K, Hardin JD, Moore KA, et al. Targeted disruption of the flk2/flt3 gene leads to deficiencies in primitive hematopoietic progenitors. Immunity. 1995;3(1):147–161.
- Vasconcelos-Dos-Santos A, Oliveira IA, Lucena MC, et al. Biosynthetic machinery involved in aberrant glycosylation: promising targets for developing of drugs against cancer. Front Oncol. 2015;5(138). doi:10.3389/fonc.2015.00138.
- Mori S, Aoyagi Y, Yanagi M, et al. Serum N-acetylglucosaminyltransferase III activities in hepatocellular carcinoma. J Gastroenterol Hepatol. 1998;13(6):610–619.
- Dube DH, Bertozzi CR. Glycans in cancer and inflammation–potential for therapeutics and diagnostics. Nat Rev Drug Discovery. 2005;4(6):477–488.
- Hauselmann I, Borsig L. Altered tumor-cell glycosylation promotes metastasis. Front Oncol. 2014;4(28). doi:10.3389/fonc.2014.00028.
- Stowell SR, Ju T, Cummings RD. Protein glycosylation in cancer. Annu Rev Pathol. 2015;10:473–510.
- Schmidt-Arras DE, Bohmer A, Markova B, et al. Tyrosine phosphorylation regulates maturation of receptor tyrosine kinases. Mol Cell Biol. 2005;25(9):3690–3703.
- Schmidt-Arras D, Bohmer SA, Koch S, et al. Anchoring of FLT3 in the endoplasmic reticulum alters signaling quality. Blood. 2009;113(15):3568–3576.
- Williams AB, Li L, Nguyen B, et al. Fluvastatin inhibits FLT3 glycosylation in human and murine cells and prolongs survival of mice with FLT3/ITD leukemia. Blood. 2012;120(15):3069–3079.
- Rosnet O, Schiff C, Pebusque MJ, et al. Human FLT3/FLK2 gene: cDNA cloning and expression in hematopoietic cells. Blood. 1993;82(4):1110–1119.
- Grassot J, Mouchiroud G, Perriere G. RTKdb: database of receptor tyrosine kinase. Nucleic Acids Res. 2003;31(1):353–358.
- van der Geer P, Hunter T, Lindberg RA. Receptor protein-tyrosine kinases and their signal transduction pathways. Annu. Rev. Cell Biol.. 1994;10:251–337.
- Griffith J, Black J, Faerman C, et al. The structural basis for autoinhibition of FLT3 by the juxtamembrane domain. Mol Cell. 2004;13(2):169–178.
- Nakao M, Yokota S, Iwai T, et al. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia. 1996;10(12):1911–1918.
- Yokota S, Kiyoi H, Nakao M, et al. Internal tandem duplication of the FLT3 gene is preferentially seen in acute myeloid leukemia and myelodysplastic syndrome among various hematological malignancies. A study on a large series of patients and cell lines. Leukemia. 1997;11(10):1605–1609.
- Frohling S, Schlenk RF, Breitruck J, et al. Prognostic significance of activating FLT3 mutations in younger adults (16 to 60 years) with acute myeloid leukemia and normal cytogenetics: a study of the AML Study Group Ulm. Blood. 2002;100(13):4372–4380.
- Abu-Duhier FM, Goodeve AC, Wilson GA, et al. Identification of novel FLT-3 Asp835 mutations in adult acute myeloid leukaemia. Br J Haematol. 2001;113(4):983–988.
- Yamamoto Y, Kiyoi H, Nakano Y, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood. 2001;97(8):2434–2439.
- Reindl C, Bagrintseva K, Vempati S, et al. Point mutations in the juxtamembrane domain of FLT3 define a new class of activating mutations in AML. Blood. 2006;107(9):3700–3707.
- Frohling S, Scholl C, Levine RL, et al. Identification of driver and passenger mutations of FLT3 by high-throughput DNA sequence analysis and functional assessment of candidate alleles. Cancer Cell. 2007;12(6):501–513.
- Breitenbuecher F, Schnittger S, Grundler R, et al. Identification of a novel type of ITD mutations located in nonjuxtamembrane domains of the FLT3 tyrosine kinase receptor. Blood. 2009;113(17):4074–4077.
- Thiede C, Steudel C, Mohr B, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002;99(12):4326–4335.
- Grundler R, Thiede C, Miething C, et al. Sensitivity toward tyrosine kinase inhibitors varies between different activating mutations of the FLT3 receptor. Blood. 2003;102(2):646–651.
- Radomska HS, Alberich-Jorda M, Will B, et al. Targeting CDK1 promotes FLT3-activated acute myeloid leukemia differentiation through C/EBPalpha. J Clin Invest. 2012;122(8):2955–2966.
- Reiter K, Polzer H, Krupka C, et al. Tyrosine kinase inhibition increases the cell surface localization of FLT3-ITD and enhances FLT3-directed immunotherapy of acute myeloid leukemia. Leukemia. 2018;32(2):313–322.
- Kayser S, Schlenk RF, Londono MC, et al. Insertion of FLT3 internal tandem duplication in the tyrosine kinase domain-1 is associated with resistance to chemotherapy and inferior outcome. Blood. 2009;114(12):2386–2392.
- Marhäll A, Heidel F, Fischer T, et al. Internal tandem duplication mutations in the tyrosine kinase domain of FLT3 display a higher oncogenic potential than the activation loop D835Y mutation. Ann Hematol. 2018;97(5):773–780.
- Cloos J, Goemans BF, Hess CJ, et al. Stability and prognostic influence of FLT3 mutations in paired initial and relapsed AML samples. Leukemia. 2006;20(7):1217–1220.
- Ozeki K, Kiyoi H, Hirose Y, et al. Biologic and clinical significance of the FLT3 transcript level in acute myeloid leukemia. Blood. 2004;103(5):1901–1908.
- Garcia JS, Stone RM. The development of FLT3 inhibitors in acute myeloid leukemia. Hematol Oncol Clin North Am. 2017;31(4):663–680.
- Zhang B, Duran PA, Piechaczyk L, et al. GRP94 rewires and buffers the FLT3-ITD signaling network and promotes survival of acute myeloid leukemic stem cells. Haematologica. 2018;104(5):e229.
- Tiong IS, Wei AH. New drugs creating new challenges in acute myeloid leukemia. Genes Chromosomes Cancer. 2019. doi:10.1002/gcc.22750.
- Jensen ON. Interpreting the protein language using proteomics. Nat Rev Mol Cell Biol. 2006;7(6):391–403.
- Helenius A, Aebi M. Intracellular functions of N-linked glycans. Science ( New York, NY). 2001;291(5512):2364–2369.
- Rudd PM, Elliott T, Cresswell P, et al. Glycosylation and the immune system. Science ( New York, NY). 2001;291(5512):2370–2376.
- Wiederschain GY. Glycobiology: progress, problems, and perspectives. Biochem Biokhimiia. 2013;78(7):679–696.
- Maeda Y, Kinoshita T. Dolichol-phosphate mannose synthase: structure, function and regulation. Biochimica et biophysica acta. 2008;1780(6):861–868.
- Larrue C, Saland E, Vergez F, et al. Antileukemic activity of 2-deoxy-d-glucose through inhibition of N-linked glycosylation in acute myeloid leukemia with FLT3-ITD or c-KIT mutations. Mol Cancer Ther. 2015;14(10):2364–2373.
- Keenan RW, Hamill RL, Occolowitz JL, et al. Biological activities of isolated tunicamycin and streptovirudin fractions. Biochemistry. 1981;20(10):2968–2973.
- Heifetz A, Keenan RW, Elbein AD. Mechanism of action of tunicamycin on the UDP-GlcNAc:dolichyl-phosphate Glc-NAc-1-phosphate transferase. Biochemistry. 1979;18(11):2186–2192.
- Lyman SD, James L, Vanden Bos T, et al. Molecular cloning of a ligand for the flt3/flk-2 tyrosine kinase receptor: a proliferative factor for primitive hematopoietic cells. Cell. 1993;75(6):1157–1167.
- Lavagna-Sevenier C, Marchetto S, Birnbaum D, et al. FLT3 signaling in hematopoietic cells involves CBL, SHC and an unknown P115 as prominent tyrosine-phosphorylated substrates. Leukemia. 1998;12(3):301–310.
- Rosnet O, Buhring HJ, deLapeyriere O, et al. Expression and signal transduction of the FLT3 tyrosine kinase receptor. Acta haematologica. 1996;95(3–4):218–223.
- Zhang S, Broxmeyer HE. Flt3 ligand induces tyrosine phosphorylation of gab1 and gab2 and their association with shp-2, grb2, and PI3 kinase. Biochem Biophys Res Commun. 2000;277(1):195–199.
- Kim KT, Baird K, Ahn JY, et al. Pim-1 is up-regulated by constitutively activated FLT3 and plays a role in FLT3-mediated cell survival. Blood. 2005;105(4):1759–1767.
- Tse KF, Mukherjee G, Small D. Constitutive activation of FLT3 stimulates multiple intracellular signal transducers and results in transformation. Leukemia. 2000;14(10):1766–1776.
- Koch S, Jacobi A, Ryser M, et al. Abnormal localization and accumulation of FLT3-ITD, a mutant receptor tyrosine kinase involved in leukemogenesis. Cells Tissues Organs (Print). 2008;188(1–2):225–235.
- Choudhary C, Olsen JV, Brandts C, et al. Mislocalized activation of oncogenic RTKs switches downstream signaling outcomes. Mol Cell. 2009;36(2):326–339.
- Mizuki M, Fenski R, Halfter H, et al. Flt3 mutations from patients with acute myeloid leukemia induce transformation of 32D cells mediated by the Ras and STAT5 pathways. Blood. 2000;96(12):3907–3914.
- Grundler R, Miething C, Thiede C, et al. FLT3-ITD and tyrosine kinase domain mutants induce 2 distinct phenotypes in a murine bone marrow transplantation model. Blood. 2005;105(12):4792–4799.
- Muller-Tidow C, Steur C, Mizuki M, et al. [Mutations of growth factor receptor Flt3 in acute myeloid leukemia: transformation of myeloid cells by Ras-dependent and Ras-independent mechanisms]. Deutsche medizinische Wochenschrift ( 1946). 2002;127(42):2195–2200.
- Choudhary C, Muller-Tidow C, Berdel WE, et al. Signal transduction of oncogenic Flt3. Int J Hematol. 2005;82(2):93–99.
- Choudhary C, Brandts C, Schwable J, et al. Activation mechanisms of STAT5 by oncogenic Flt3-ITD. Blood. 2007;110(1):370–374.
- Murata K, Kumagai H, Kawashima T, et al. Selective cytotoxic mechanism of GTP-14564, a novel tyrosine kinase inhibitor in leukemia cells expressing a constitutively active Fms-like tyrosine kinase 3 (FLT3). J Biol Chem. 2003;278(35):32892–32898.
- Sallmyr A, Fan J, Datta K, et al. Internal tandem duplication of FLT3 (FLT3/ITD) induces increased ROS production, DNA damage, and misrepair: implications for poor prognosis in AML. Blood. 2008;111(6):3173–3182.
- Brown J. Effects of 2-deoxyglucose on carbohydrate metablism: review of the literature and studies in the rat. Metab Clin Exp. 1962;11:1098–1112.
- Kang HT, Hwang ES. 2-Deoxyglucose: an anticancer and antiviral therapeutic, but not any more a low glucose mimetic. Life Sci. 2006;78(12):1392–1399.
- Pelicano H, Martin DS, Xu RH, et al. Glycolysis inhibition for anticancer treatment. Oncogene. 2006;25(34):4633–4646.
- Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol. 2003;4(3):181–191.
- Parodi AJ. Protein glucosylation and its role in protein folding. Annu Rev Biochem. 2000;69:69–93.
- Andresen L, Skovbakke SL, Persson G, et al. 2-deoxy D-glucose prevents cell surface expression of NKG2D ligands through inhibition of N-linked glycosylation. J Immunol ( Baltimore, Md: 1950). 2012;188(4):1847–1855.
- Kurtoglu M, Gao N, Shang J, et al. Under normoxia, 2-deoxy-D-glucose elicits cell death in select tumor types not by inhibition of glycolysis but by interfering with N-linked glycosylation. Mol Cancer Ther. 2007;6(11):3049–3058.
- Xi H, Kurtoglu M, Liu H, et al. 2-Deoxy-D-glucose activates autophagy via endoplasmic reticulum stress rather than ATP depletion. Cancer Chemother Pharmacol. 2011;67(4):899–910.
- Kang HT, Ju JW, Cho JW, et al. Down-regulation of Sp1 activity through modulation of O-glycosylation by treatment with a low glucose mimetic, 2-deoxyglucose. J Biol Chem. 2003;278(51):51223–51231.
- Han X, Zhang X, Li H, et al. Tunicamycin enhances the antitumor activity of Trastuzumab on breast cancer in vitro and in vivo. Oncotarget. 2015;6(36):38912–38925.
- Contessa JN, Bhojani MS, Freeze HH, et al. Molecular imaging of N-linked glycosylation suggests glycan biosynthesis is a novel target for cancer therapy. Clin Cancer Research: Official J Am Assoc Cancer Res. 2010;16(12):3205–3214.
- Contessa JN, Bhojani MS, Freeze HH, et al. Inhibition of N-linked glycosylation disrupts receptor tyrosine kinase signaling in tumor cells. Cancer Res. 2008;68(10):3803–3809.
- Ahsan A, Hiniker SM, Ramanand SG, et al. Role of epidermal growth factor receptor degradation in cisplatin-induced cytotoxicity in head and neck cancer. Cancer Res. 2010;70(7):2862–2869.
- Helenius A, Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem. 2004;73:1019–1049.
- Ahmmed B, Khan MN, Nisar MA, et al. Tunicamycin enhances the suppressive effects of cisplatin on lung cancer growth through PTX3 glycosylation via AKT/NF-kappaB signaling pathway. Int J Oncol. 2019;54(2):431–442.
- Tsitsipatis D, Jayavelu AK, Muller JP, et al. Synergistic killing of FLT3ITD-positive AML cells by combined inhibition of tyrosine-kinase activity and N-glycosylation. Oncotarget. 2017;8(16):26613–26624.
- Corsini A, Maggi FM, Catapano AL. Pharmacology of competitive inhibitors of HMG-CoA reductase. Pharmacol Res. 1995;31(1):9–27.
- Mo H, Elson CE. Studies of the isoprenoid-mediated inhibition of mevalonate synthesis applied to cancer chemotherapy and chemoprevention. Exp Biol Med ( Maywood. NJ). 2004;229(7):567–585.
- Stirewalt DL, Appelbaum FR, Willman CL, et al. Mevastatin can increase toxicity in primary AMLs exposed to standard therapeutic agents, but statin efficacy is not simply associated with ras hotspot mutations or overexpression. Leuk Res. 2003;27(2):133–145.
- Lee J, Lee I, Han B, et al. Effect of simvastatin on cetuximab resistance in human colorectal cancer with KRAS mutations. J Natl Cancer Inst. 2011;103(8):674–688.
- Martirosyan A, Clendening JW, Goard CA, et al. Lovastatin induces apoptosis of ovarian cancer cells and synergizes with doxorubicin: potential therapeutic relevance. BMC Cancer. 2010;10(103). doi:10.1186/1471-2407-10-103.
- Holstein SA, Knapp HR, Clamon GH, et al. Pharmacodynamic effects of high dose lovastatin in subjects with advanced malignancies. Cancer Chemother Pharmacol. 2006;57(2):155–164.
- Hamadmad SN, Hohl RJ. Lovastatin suppresses erythropoietin receptor surface expression through dual inhibition of glycosylation and geranylgeranylation. Biochem Pharmacol. 2007;74(4):590–600.
- Yoshimura A, D’Andrea AD, Lodish HF. Friend spleen focus-forming virus glycoprotein gp55 interacts with the erythropoietin receptor in the endoplasmic reticulum and affects receptor metabolism. Proc Natl Acad Sci U.S.A. 1990;87(11):4139–4143.
- Siddals KW, Marshman E, Westwood M, et al. Abrogation of insulin-like growth factor-I (IGF-I) and insulin action by mevalonic acid depletion: synergy between protein prenylation and receptor glycosylation pathways. J Biol Chem. 2004;279(37):38353–38359.
- Tahvanainen J, Kylaniemi MK, Kanduri K, et al. Proviral integration site for moloney murine leukemia virus (PIM) kinases promote human T helper 1 cell differentiation. J Biol Chem. 2013;288(5):3048–3058.
- Xie Y, Burcu M, Linn DE, et al. Pim-1 kinase protects P-glycoprotein from degradation and enables its glycosylation and cell surface expression. Mol Pharmacol. 2010;78(2):310–318.
- Bullock AN, Debreczeni J, Amos AL, et al. Structure and substrate specificity of the Pim-1 kinase. J Biol Chem. 2005;280(50):41675–41682.
- Kamemura K, Hart GW. Dynamic interplay between O-glycosylation and O-phosphorylation of nucleocytoplasmic proteins: a new paradigm for metabolic control of signal transduction and transcription. Prog Nucleic Acid Res Mol Biol. 2003;73:107–136.
- Natarajan K, Xie Y, Burcu M, et al. Pim-1 kinase phosphorylates and stabilizes 130 kDa FLT3 and promotes aberrant STAT5 signaling in acute myeloid leukemia with FLT3 internal tandem duplication. PloS one. 2013;8(9):e74653.
- Marzec M, Eletto D, Argon Y. GRP94: an HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochimica et biophysica acta. 2012;1823(3):774–787.
- Rutherford SL, Lindquist S. Hsp90 as a capacitor for morphological evolution. Nature. 1998;396(6709):336–342.
- Trepel J, Mollapour M, Giaccone G, et al. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer. 2010;10(8):537–549.
- Minami Y, Kiyoi H, Yamamoto Y, et al. Selective apoptosis of tandemly duplicated FLT3-transformed leukemia cells by Hsp90 inhibitors. Leukemia. 2002;16(8):1535–1540.
- Taldone T, Patel PD, Patel M, et al. Experimental and structural testing module to analyze paralogue-specificity and affinity in the Hsp90 inhibitors series. J Med Chem. 2013;56(17):6803–6818.
- Weisberg E, Boulton C, Kelly LM, et al. Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell. 2002;1(5):433–443.
- Wolfert MA, Boons GJ. Adaptive immune activation: glycosylation does matter. Nat Chem Biol. 2013;9(12):776–784.
- von Bubnoff N, Engh RA, Aberg E, et al. FMS-like tyrosine kinase 3-internal tandem duplication tyrosine kinase inhibitors display a nonoverlapping profile of resistance mutations in vitro. Cancer Res. 2009;69(7):3032–3041.