
ABSTRACT
Objectives
β-Thalassemia (β-thal) is a genetic disease of the blood caused by mutations in the β-globin gene. Conventional methods for detecting thalassemia variants often miss rare and novel variants. Identifying the rare and novel β-thal variants, especially in the high prevalence regions, would enable better disease prevention.
Methods
A Chinese family who had joined the Thalassemia Prevention Program was recruited in this study. The β-thal carrier screening was performed using next-generation sequencing (NGS), and the results were validated through direct DNA sequencing. Hematological parameters were analyzed, and hemoglobin electrophoresis was performed. Additionally, the presence of thalassemia-associated deletions was determined using gap-polymerase chain reaction.
Results
A novel frameshift variant of β-thal, HBB:c.14delC(Codon 4, -C), was identified in a 31-year-old Chinese man. Subsequent genetic investigation showed that his mother also carried this novel variant. Hematological analysis and clinical evaluation suggested that this variant was present in the heterozygous state and might belong to a severe phenotype of β-thal.
Conclusions
We identified a novel frameshift variant of β-thal. NGS has the potential for identifying rare and novel thalassemia variants and broadening the spectrum of thalassemia screening and thus may contribute to effective prevention of thalassemia.
Introduction
Thalassemia is a common genetic disease caused by disorders in the synthesis of hemoglobin. In thalassemia, patients have defects in either the α or β globin chain, resulting in abnormal red blood cells. Based on the genes that are affected, thalassemia is classified into α-thalassemia (α-Thal) and β-thal [Citation1]. More than 900 genomic alterations of β-globin and over 300 variants of the β-globin gene resulting in β-thal have been reported worldwide [Citation2]. β-thal, according to its clinical severity, has been classified into β+ and β0 [Citation3]. A genetic screening, using a reverse dot-blot hybridization (RDB) method for 17 regular variants of the β-globin gene, is routinely carried out in China [Citation4]. This genetic screen combined with hematological indices can identify most β-thal carriers [Citation5]. In addition, gap-polymerase chain reaction (Gap-PCR), high resolution melting analysis, multiplex ligation-dependent probe amplification (MLPA), and Sanger sequencing are used to identify subjects with significant β-thal traits [Citation6–8]. Several rare and novel variants of β-thal are being reported in recent years. In the Chinese population, about 50 different variants of β-thal have been reported, and this number might further increase in the near future [Citation3, Citation9–11]. Faster and efficient screening strategies would enable better identification of the new variants. Next-generation sequencing (NGS), with its high sensitivity and specificity, is regarded as a competitive screening method that could improve thalassemia carrier screening in a high prevalence population, and provide a comprehensive assessment of the current thalassemia screening strategies [Citation12].
Since 2019, the Thalassemia Prevention Program, sponsored by the Local Government, has used NGS for the detection of β-thal. Through this screening program, we identified a novel frameshift variant in a 31-year-old Chinese man, who presented with hematological parameters indicating typical β-thal. This novel variant in a heterozygous state at codon 4 of the β-globin gene [HBB:c.14delC(Codon 4, -C)] was confirmed by Sanger sequencing. The novel frameshift variant was also found in the proband’s mother, who also showed typical traits of β-thal. This β-thal variant that caused a microcytic, hypochromic anemia phenotype has never been reported before.
Materials and methods
Subjects
The members of the Chinese family, reported in the study, were participants of the Government Thalassemia Prevention Program. The proband is a 31-year-old man of Han ethnicity from Ganzhou City, Jiangxi Province, Southeast China. The proband and his family underwent hematological screening and hemoglobin electrophoresis. Peripheral blood samples from the family were collected and stored for further investigation. No history of blood transfusion was reported by the family members. Written informed consent was obtained from the study participants.
Molecular analysis
The three most common α-thalassemia-associated deletions (–SEA, -α3.7, and -α4.2), two rare deletions (–FIL and –THAI), and two common β-globin gene deletions (SEA-HPFH and Gγ+(Asγδβ)) in China were characterized using Gap-PCR (BGI Biological Technology, Shenzhen).
Full-length HBA1, HBA2, and HBB were amplified by PCR. The amplicons spanned all the exons and introns of HBA1, HBA2, and HBB genes, which ensured that most thalassemia-associated variants and CNVs in the HbVar database could be detected. Sequencing libraries were constructed according to the Illumina HiSeq Sequencing Library Preparation Protocol. These libraries were further paired-end-sequenced for 100 base pairs (PE100) using an Illumina HiSeq2000 machine. The protocol for the bioinformatic analysis and identification of hemoglobin gene variants was described previously [Citation13].
Sanger sequencing of the β-globin gene was performed for validating the variants. We designed specific primers according to the known DNA sequences around the HBB gene to perform PCR. The primers were as follows: 5′- TGGAGCCACACCCTAGG-3′ as the forward primer and 5′- TGAAGTTCTCAGGATCCACGT-3′ as the reverse primer. All primers were synthesized by Invitrogen (Shanghai).
Results
The hematological parameters of the proband and his family members are shown in . We suspected that the proband and his mother could be β-thalassemia carriers based on the levels of the decreased MCV and MCH and the increased HbA2. In addition, his wife and father were considered not to be affected based on the normal levels of MCV, MCH, and HbA2. We did not observe the presence of common large deletions in β-globin gene. The results of NGS indicated that there was a frameshift variant existing in the proband . As shown in , a novel variant, HBB:c.14delC (Codon 4, -C), was validated in the proband by Sanger sequencing. The proband carried a deletion of a single C base at nucleotide position 14 from the initiation codon ATG in the 5’translated region of the human β-globin gene and the frameshift change results in a premature stop codon at codon18 that has never been reported before. The proband’s mother, who also presented with abnormal levels of HbA2, MCV, and MCH, carried the same variant. Based on this, the thalassemia genotype for the proband and his mother can be described as αα/αα, βCD4M/βN.
Figure 1. The results of Next-generation sequencing in the proband. The β-globin gene sequence highlighted in the red box indicated that there was a frameshift peak and the proband carried a mutation: HBB:c.14delC. The reference sequence is included in the figure.
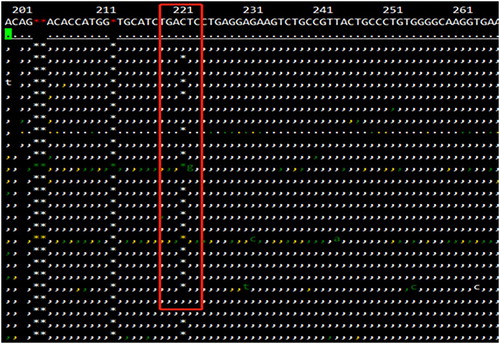
Figure 2. The results of the variant confirmed by Sanger sequence and its effect for the transcription process of β-globin gene. (A) Forward and reverse sequence data of the β-globin gene confirms the codon 4 deletion mutation in the heterozygous state and the deletion results in numerous ambiguities (peaks overlapping). (B) Nucleotide sequence of the β-globin gene on exon 1, codons 3–19. Note the frameshift change starting at codon 4 that results in a premature stop codon at position 18.
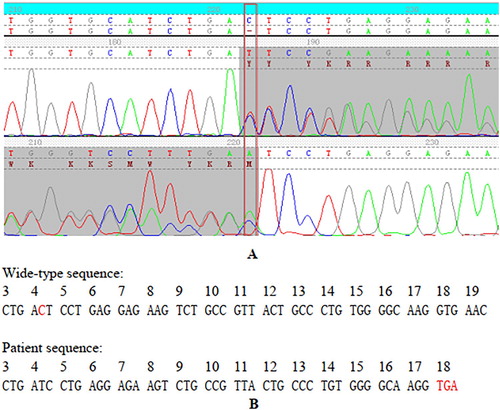
Table 1. Hematological parameters in the family with a frameshift mutation at Codon 4 (-C) in the β-globin gene.
Discussion
β-thal, one of the most prevalent hereditary diseases around the world, is caused by reduced or abolished β-globin chain synthesis due to the presence of variants of the β-globin gene. It is common in people of Mediterranean countries, the Middle East, the Indian subcontinent, Southeast Asia, and Southern China [Citation14–16]. The rate of β-thal in the city of Ganzhou, located in the southern Jiangxi Province, has been reported to be 2.3% [Citation17]. The identification of new β-thalassemia variants from this region is necessary for better management of thalassemia prevention. In the routine screening program, RDB is used widely to detect the 17 common β-thal variants [Citation18]. However, there exists the possibility of missing rare and novel variants; this is especially crucial when one person is confirmed to be afflicted with thalassemia, and their partner is a suspected carrier [Citation19]. Therefore, NGS should be applied to screen for β-globin gene variants for improved sensitivity and specificity. In addition, NGS has a substantial advantage of being time- and cost-efficient compared to traditional methods [Citation20].
In this study, we reported a novel frameshift variant of β-thalassemia, a deletion at codon 4 of the β-globin gene (HBB: c.14delC) detected during the process of carrier screening via NGS. Subsequently, Sanger sequencing was performed to validate the results. The previously reported variants within codon 4 included nucleotide substitutions such as A to C [ACT > CCT, HBB:c.13A > C] and C to A [ACT > AAT, HBB:c.14C > A] [Citation21]. These variants have a normal clinical presentation with about 40% Hb X in total Hb. So far, there has been no report of a missing nucleotide at codon 4, according to the human HbVar database (http://globin.cse.psu.edu). About ten single nucleotide deletions have been reported in exon 1 of the β-globin gene. Several nucleotide deletion variants described in codons 1, 2, 5, and 6 of the β-globin gene are responsible for β0-thalassemic alleles [Citation22–25].
In the proband’s family, the novel frameshift variant results in premature termination of translation due to the premature stop codon UGA at codon 18. The novel variant was predicted to be pathogenic by at least two of the in-silico prediction tools. The combined data from clinical evaluation, hematological analysis, and sequencing revealed that the novel β-thal variant – deletion of a single C base at codon 4 (HBB: c.14delC) – might belong to the severe β-thal class and that the variant existed in a heterozygotic state in the family. In the heterozygous state, the variant carriers have no β chain synthesized from the abnormal allele, while all the β chain was synthesized from the normal allele and presented the phenotype of a typical heterozygous β-thal. Thus, the patient and his mother’s eventual genotype were αα/αα, βCD4M/βN based on the results from Gap-PCR.
In summary, We identified a novel frameshift variant of β-thal, HBB:c.14delC (Codon 4, -C), through the Thalassemia Prevention Program, using NGS. Our report suggests that NGS may expand the spectrum of thalassemia and enable thalassemia prevention more feasible without rare or novel variants missed.
Acknowledgements
The authors would like to thank the family members in the study.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Notes
RBC: red blood cell count; Hb: hemoglobin; MCV: mean corpuscular volume; MCH: mean corpuscular Hb.
References
- Zhuang J, Zheng Y, Wang Y, et al. Identification of a new β-thalassaemia variant Term CD+32(HBB: c.32A > C) in two Chinese families. J Clin Pathol. 2020; 73(9):1–4.
- Chen JK, Xin XQ, Huang JG. A novel β-thalassemia mutation in a Chinese family: IVS-II-203-205 (TCT > CC) (HBB: c.315 + 203TCT > CC). Hemoglobin. 2018;42(3):159–160.
- Jia W, Wang W, Zhu H, et al. A novel mutation at HBB: c.91delA (codon 30, -A) causing β-thalassemia in a Chinese family. Acta Haematol. 2019;142(4):249–252.
- Liu X, Su L, Li G, et al. Analysis of β -thalassemia mutations in Guizhou Province. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2014;31(5):561–564.
- Pornprasert S, Tookjai M, Punyamung M, et al. Proficiency testing program for hemoglobin E, A2 and F analysis in Thailand using lyophilized hemoglobin control materials. Clin Chem Lab Med. 2018;56(4):602–608.
- Zhang J, Yang Y, Li P, et al. Analysis of deletional hereditary persistence of fetal hemoglobin/δβ-thalassemia and δ-globin gene mutations in Southwestern China. Mol Genet Genom Med. 2019;7(6):e706.
- He S, Wei Y, Lin L, et al. The prevalence and molecular characterization of (δβ) -thalassemia and hereditary persistence of fetal hemoglobin in the Chinese Zhuang population. J Clin Lab Anal. 2018;323(3):e22304.
- Islam MT, Sarkar SK, Sultana N, et al. High resolution melting curve analysis targeting the HBB gene mutational hot-spot offers a reliable screening approach for all common as well as most of the rare beta-globin gene mutations in Bangladesh. BMC Genet. 2018;19(1):1.
- Li DZ, Liao C, Li J, et al. A novel beta-globin gene deletion (codons 89-93) in a Chinese family. Ann Hematol. 2010;89(3):323–325.
- Huang H, Xu L, Lin N, et al. A new β-thalassemia deletion mutation [codon 36 (-C)] observed in a Chinese woman. Hemoglobin. 2010;34(6):599–603.
- Yi P, Yu F, Huang S, et al. Identification of a novel frameshift mutation at codon 53 (-T) in the beta-globin gene causing dominantly inherited beta-thalassemia in a Chinese Miao family. Blood Cells Mol Dis. 2008;41(1):56–59.
- Zhang H, Li C, Li J, et al. Next-generation sequencing improves molecular epidemiological characterization of thalassemia in Chenzhou region, P.R. China. J Clin Lab Anal. 2019;33(4):e22845.
- He J, Song W, Yang J, et al. Next-generation sequencing improves thalassemia carrier screening among premarital adults in a high prevalence population: the Dai nationality, China. Genet Med. 2017;19(9):1022–1031.
- Rund D, Rachmilewitz E. Beta-thalassemia. N Engl J Med. 2005;353(11):1135–1146.
- Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86(6):480–487.
- Yang Z, Cui Q, Zhou W, et al. Comparison of gene mutation spectrum of thalassemia in different regions of China and Southeast Asia. Mol Genet Genom Med. 2019;7(6):e680.
- Lin M, Zhong TY, Chen YG, et al. Molecular epidemiological characterization and health burden of thalassemia in Jiangxi Province, P. R. China. PLoS One. 2014;9(7):e101505.
- Charoenkwan P, Sirichotiyakul S, Phusua A, et al. High-resolution melting analysis for prenatal diagnosis of beta-thalassemia in northern Thailand. Int J Hematol. 2017;106(6):757–764.
- Suwannakhon N, Pongsawatkul K, Seeratanachot T, et al. The shortcut strategy for beta thalassemia prevention. Hematol Rep. 2018;10(2):7530.
- Shang X, Peng Z, Ye Y, et al. Rapid targeted next-generation sequencing Platform for molecular screening and clinical genotyping in subjects with hemoglobinopathies. EBioMedicine. 2017;23:150–159.
- Bissé E, Zorn N, Boussert S, et al. Characterization of hemoglobin Würzburg (alpha2beta2 4(A1)Thr–>Asn), a new electrophoretically silent variant, by mass spectrometry and molecular modeling studies. J Chromatogr A. 2006;1115(1-2):118–124.
- Rosatelli MC, Dozy A, Faà V, et al. Molecular characterization of beta-thalassemia in the Sardinian population. Am J Hum Genet. 1992;50(2):422–426.
- Nagar R, Sinha S, Raman R. Genotype-phenotype correlation and report of novel mutations in β-globin gene in thalassemia patients. Blood Cells Mol Dis. 2015;55(1):10–14.
- Gonzalez-Redondo JM, Stoming TA, Lanclos KD, et al. Clinical and genetic heterogeneity in black patients with homozygous beta-thalassemia from the southeastern United States. Blood. 1988;72(3):1007–1014.
- Kollia P, Gonzalez-Redondo JM, Stoming TA, et al. Frameshift codon 5 [Fsc-5 (-CT)] thalassemia; a novel mutation detected in a Greek patient. Hemoglobin. 1989;13(6):597–604.