
ABSTRACT
Background
: Congenital dysfibrinogenemia (CD) is a coagulation disorder caused by mutations in the fibrinogen genes, which result in abnormal fibrinogen function. However, the precise pathogenesis underlying it remains unclear.
Methods
: In this study, we identified a novel heterozygous mutation in an asymptomatic patient with CD caused by γ Ala327Val mutation. Aimed to investigate the pathogenesis, functional studies of fibrinogen isolated from the proband and her family members were performed, such as coagulation function, fibrinogen aggregation test, and fibrin clot lysis test. Coagulation was monitored using a thromboelastometer, and the fibrin clot network structure was observed by scanning electron microscopy. The effect of the mutation on fibrinogen structure and function was predicted by molecular modeling.
Results
: The fibrinogen activity concentration in patients with CD was significantly lower than that in healthy individuals, indicating that fibrinogen activity was low. Proband’s fibrinogen activity concentration was 0.75 g/L(Clauss method) and antigen concentration (immune turbidimetry method) was 1.59 g/L(normal reference range for both parameters: 2.0–4.0 g/L). Thromboelastography showed that the K value of patients with CD was higher than that of healthy individuals and Angle values were decreased, indicating that mutation impaired fibrinogen function. Compared to fibrinogen from healthy individuals, fiber network structure of the proband was loose, pore size was increased, and fiber branch nodes were increased.
Conclusions
: Ala327Val heterozygous missense mutation leads to changes in the structure of fibrinogen D region and impairs the aggregation function of fibrinogen. This mutation is reported here for the first time.
1. Introduction
Fibrinogen (Fg), also known as coagulation factor I, is mainly produced by liver cells and is secreted into peripheral blood. Its plasma concentration is 2–4 g/L [Citation1]. Fibrinogen is involved in the formation of fibrin clots and platelet aggregation, and plays an important role in hemostasis. The relative molecular weight of fibrinogen is 340 kD; it is composed of two identical subunits, each of which comprises three different polypeptide chains (A α, B β, and γ); these two subunits are linked by disulfide bonds [Citation2]. Fibrinogen molecules have three major functional structures, the central nodule (E-region) that contains the N-terminus of each chain and two symmetric distal spherical D-regions that contain the C-terminus of Bβ- andγ-chains. The three polypeptide chains A α, B β, and γ are encoded by FGA, FGB, and FGG genes (on chromosome 4), respectively [Citation3].
Mutations in FGA, FGB, or FGG may lead to the development of congenital dysfibrinogenemia (CD). CD is a congenital blood disease caused by defects in fibrinogen genes, which lead to abnormal structure and function of fibrinogen molecules, and may affect coagulation. The clinical manifestations of CD are diverse; most patients with CD are asymptomatic [Citation4], but a small number of patients with CD have thrombosis [Citation5, Citation6] and bleeding events, and some patients have pulmonary hypertension and other symptoms [Citation7]. According to the latest published study, bleeding was present in 42% of patients with dysfibrinogenemia [Citation8].
In this study, we identified a novel heterozygous mutation in an asymptomatic patient with congenital dysfibrinogenemia with the amino acid substitution of γAla327Val. After the approval of the hospital ethics committee and the informed consent of patients, functional studies of fibrinogen isolated from the proband and her family members were performed to study the molecular pathogenesis of CD caused by γ Ala327Val heterozygous missense mutation.
2. Methods
2.1. Basic data of patients and routine examination of coagulation function
The proband was a 45-year-old female patient. When she came to the hospital for routine physical examination during pregnancy, her coagulation function test result was abnormal: fibrinogen activity concentration was 0.75 g/L (Clauss method) and antigen concentration (immune turbidimetry method) was 1.59 g/L (normal reference range for both parameters: 2.0–4.0 g/L). Fibrinogen activity concentration was significantly lower than fibrinogen antigen concentration. Liver function, kidney function, and blood routine tests were normal. So the patient was initially diagnosed with dysfibrinogenemia. There were no bleeding or thrombotic events in daily life. The proband was pregnancy in 2006 and delivered by caesarean section. At that time, fibrinogen levels of proband was low but the caesarean section was smooth. Four members of her family were examined for coagulation function and other parameters.
DNA was extracted from the peripheral blood samples of the proband and four members of her family. DNA sequencing was performed by Beijing Liuhe Huada Gene Technology Co., Ltd, Beijing, China.
2.2. Fibrinogen aggregation test
Venous blood (5 ml, in sodium citrate anticoagulant) was drawn from the patients and healthy individuals, and centrifuged at 4°C, 3000 rpm, for 15 min to separate the plasma. Then, 1 ml sodium citrate (0.09 mol/L) and 666 μl saturated ammonium sulfate (pH 5.5) were added to 1 ml plasma, and the mixture was incubated at 25°C for 70 min. After centrifugation, the supernatant was discarded and fibrinogen was obtained. Fibrinogen was purified by precipitation with 20%saturated ammonium sulfate, and the pellet was washed 3 times with 25% saturated ammonium sulfate [Citation9]. The reaction system of the fibrinogen aggregation test was as follows: Fibrinogen 0.5 mg/ml (final concentration), 5 ml of 2 mol/L NaCl, 5 ml of 2 mmol/L CaCl2, and 20 mmol/L HEPES buffer to make the volume up to 90 ml. Then, 10 U/ml of 10 μl thrombin was added and the optical density (OD) of the sample at 365 nm was continuously monitored (one reading every 20 s for 30 min) using a multimode plate reader Multiskan Go (ThermoFischer). The aggregation curve was drawn according to the time and the corresponding OD value.
2.3. Fibrin clot dissolution test
Fibrin clot dissolution test was performed by adding fibrinolytic enzyme and tPA into the fibrinogen aggregation test system to activate fibrin clot dissolution, and we continuously measured the OD value of the reaction system to reflect the rate of fibrin clot dissolution. The reaction system used for this test was as follows: The final concentrations of fibrinogen, thrombin, plasminogen, tPA, CaCl2, and NaCl were 0.5 mg/ml, 0.5 U/ml, 0.12 U/ml, 0.1 mg/ml, 8 mmol/L, 0.12 mmol/L, and 20 mmol/L, respectively. Finally, 20 mmol/L HEPES was used to make up the volume of the reaction system to 200 ml. The OD values of the samples at 365 nm were continuously monitored with Multiskan Go (ThermoFisher, U.S.A.) for 30 min at 20 s intervals. The fibrin clot dissolution curve was drawn according to the time and the corresponding OD value.
2.4. Thromboelastography
Whole blood samples (1 ml) from the study subjects were added to the reagent bottle (Shaanxi Yuze Yi Medical Technology Co., Ltd Shaanxi, China); Then, 340 µl of the sample with 20 μl of 0.2 mol/L CaCl2 was added into the detection cup of the thromboelastography (TEG) instrument (Shaanxi Yuze Yi Medical Technology Co., Ltd, Shaanxi, China). As fibrinogen began to aggregate, the probe placed in the detection cup was subjected to shear stress produced during the formation and dissolution of the blood clot. The generated signal was transmitted to the processor in the form of a current and a TEG curve was obtained.
2.5. Scanning electron microscopy
The proband and healthy individuals were administered 33 μl of fibrinogen each, and thrombin was added at a final concentration of 2 U/ml. After incubation for 3 h at 37°C, the samples from proband and healthy individuals were rinsed with PBS buffer (pH 7.4, 0.1 mol/L), and incubated with 3% glutaraldehyde for 2 h. Glutaraldehyde was discarded, samples were rinsed with PBS solution, and dehydrated with alcohol. The ultrastructure of the fibrin clot was observed with VEGA3LMU scanning electron microscope (TESCAN, Czech Republic).
2.6. Modeling and analysis of amino acid mutation
The amino acid sequence of fibrinogen was obtained from the NCBI database, and a model of the fibrinogen structure was constructed using SWISS-MODEL website (https://swissmodel.expasy.org/)/. Swiss-Pdb Viewer software was used to analyze the effect of γ Ala327Val mutation on the function of fibrinogen.
3. Results
3.1. Routine coagulation function tests
The Results of the laboratory examination for the proband including electrolyte, liver and kidney function, and blood routine tests were normal. Examination of these parameters for other members of the family showed that the coagulation results of the proband’s sister, two brothers, and proband’s daughter were similar to those of the proband. The results of the coagulation function test are given in . A single base heterozygous missense mutation, FGG c.1058C > T, p.Ala327Val (Ala353Val in mature protein chain), in exon 8 of the fibrinogen gene was found in the proband and her family members ().
Figure 1. Sequences of gene.1: Proband; 2-5: Proband’s family member; 6: Healthy individual (↓: FGG gene c.1058C>T heterozygous mutation).
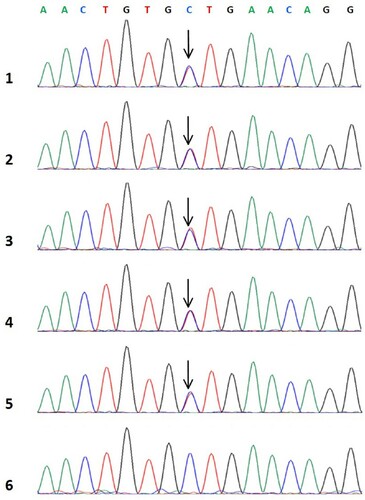
Table 1. Coagulation function of proband and her family members.
3.2. Fibrinogen aggregation test
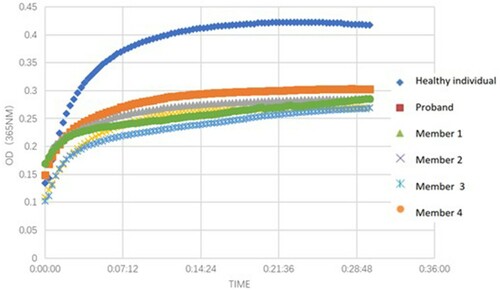
Compared with the healthy individuals, the OD values of fibrinogen aggregation curves for fibrinogen isolated from the proband and her family members were changed slightly, and the maximum OD values from 5 patients (average 0.283) were lower than those of the healthy individual (0.422) ().
Figure 2. Fibrin polymerization curves.
3.3. Fibrin clot dissolution test
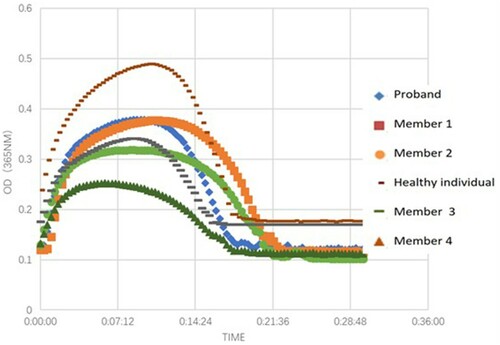
The clot turbidity began to decrease after 496 s on average (healthy individuals: after 660 s). There was no fibrinolysis delay and resistance during clot dissolution ().
Figure 3. Fibrin clot lysis curves.
3.4. Thromboelastography
The K values (time when the strength of blood clot reaches 20 mm) of patients with CD were increased; the average value was 3.7 min for the patients whereas it was 2.3 min for healthy individuals. Further, the Angle values (the angle between tangent line and horizontal line from the point of clot formation to the maximum curve radian of the figure) were decreased. The average value was 52.8° for the patients, whereas it was 61.5° for the healthy individuals. The thromboelastography results are shown in and .
Figure 4. Thromboelastography. 1: Healthy individual; 2-6: Proband and her family members.
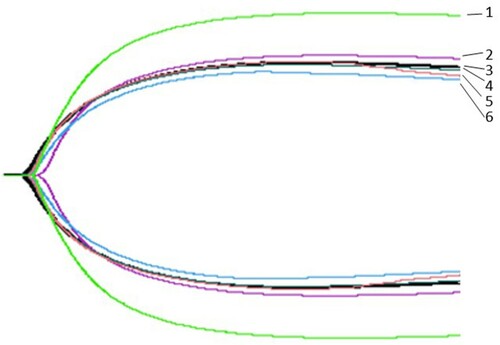
Table 2. Thromboelastography in the proband, her family members, and healthy individual.
3.5. Ultrastructure of fibrin clot
Shown in (A) is the fibrin clot structure of the healthy individual under a scanning electron microscope. The diameter of the fiber filament was uniform, the fiber nodes were few, the arrangement was neat and compact, the fiber filament interweaved and overlapped to form a fiber network, the spatial structure was relatively dense, and the network pore size was small. (B) The fibrin clot network structure of the proband showed that the fiber filaments were of different thickness, arranged irregularly, and the ends of the fiber filaments were curled into a mass. The fiber network space structure was loose, the network aperture increased, and the fiber branch nodes were more than those in the healthy individual.
Figure 5. Scanning electron microscopy of fibrin clot. A: Healthy individual; B: Proband.

3.6. Modeling and analysis of amino acid mutations
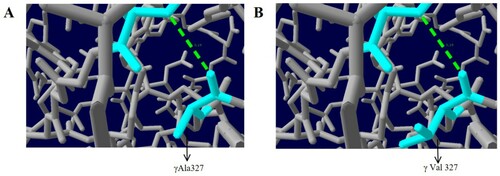
In the molecular model of wild-type fibrinogen, Ala327 is located in the α helix of D region of the fibrinogen γ chain. The α helix is crucial to maintaining protein stability. The γ Ala327 backbone and γ Ser332 backbone form a hydrogen bond, which is 3.19 nm long. When Ala is replaced by Val, the hydrogen bond between γ-Val327 and γ-Ser332 did not change, but the side chain became longer, which affected its spatial structure, and the electrostatic force around it changed, which led to a change in the structure of the α-helix and weakening of protein stability ().
Figure 6. Analysis of the γAla327Val mutation with protein modelling. A: Healthy individual; B: Proband.
4. Discussion
Congenital dysfibrinogenemia (CD) is a hereditary blood disease caused by defects in fibrinogen genes that lead to an abnormal structure and function of fibrinogen. The mutations in the three fibrinogen genes include missense mutations caused by single base substitutions, frameshift mutation caused by single base insertion or deletion, and mutation in the regulatory region. So far, over 450 mutations in fibrinogen genes have been registered (www.geht.org). Single amino acid mutations are reported at 212 sites, of which mutations in the FGA gene are the most common, followed by mutations in the FGG [Citation10]. In fact, dysfibrinogenemia is almost always caused by heterozygous missense mutations (99.3%) in FGA and FGG [Citation11].
In this study, the genetic testing of proband and her family members revealed a heterozygous missense mutation (C. 1058C>T, p.Ala327Val) in exon 8 of the FGG gene, which resulted in the substitution of alanine with valine at position 327 of the γ chain. Structure modeling analysis showed that this caused a change in the spatial position and electrostatic force of fibrinogen, resulting in a different structure of the α-helix affecting its stability.
Although the interaction between the D:E regions is the main driving force of fibrinogen aggregation, D:D interaction is important for fibrin oligomer formation [Citation12–14]. The role of D:D interaction is to guide the D regions of the two fibrinogen monomers to connect. This D:D interaction assists γ chain polymerization, which is necessary for end-to-end fibrin binding. By projecting the D:D region and analyzing its crystal structure, it was found that the interface of D:D polymerization is composed of hydrogen bonds between two adjacent fibrinogen molecules and that D:D interaction is critical for the horizontal and linear aggregation of fibrin monomers [Citation12, Citation15]. The γC region (residues 143–411) forms a single globular domain (D regions) and plays a critical role in fibrinogen assembly and secretion [Citation16]. The mutation site γAla327 investigated in this study is located in this region. In γAla327Val, alanine is replaced by valine, which affects the secondary structure, changes the spatial arrangement that consecutively impairs D:D binding between the γ chains of fibrinogen, and hinders the formation of fibrils, thereby affecting the aggregation of fibrinogen. Our aggregation test and thromboelastography results confirmed that the proband and her family members had impaired fibrin aggregation.
We investigated the ultrastructure of the probands’ fibrin clots using scanning electron microscopy. The filaments showed different sizes from what is seen in regular clot formation, their arrangement was irregular, and their ends were coiled. Furthermore, the fiber network was loose, and its pores were enlarged, and there were many fiber nodes. Impaired D:D interaction between adjacent fibrinogen molecules frequently leads to the lateral extension of the filament, increasing the number of branch points and forming thinner filaments than what is seen in normal clot formation [Citation17]. The substitution of γAla327 causes incorrect end-to-end orientation of fibrin monomers during aggregation, resulting in the increase in fiber branching. Moreover, normal fibrin monomers can combine with mutant monomers during polymerization, resulting in variable fiber diameter, irregular arrangement, and a loose and large mesh-like structure of the fiber network. An analysis of the mutant fibrin clot network showed that the fibrin clots with impaired D:D interaction (γArg275His, γArg275Cys and γArg375Gly) had more pores in the network structure, and that the network comprised many conical fibers and pores [Citation18]. These results are similar to those of in this study. Sugo et al [Citation18] suggested that the structure of fibrin clots might be related to the clinical CD phenotype and that the phenotype could be predicted by studying the ultrastructure of the fibrin clot. Studies have shown that the dissolution rate of fibrin clots depends on the density of their structure rather than the diameter of the fibrin fibers and that loose networks composed of crude fibers decompose more easily than the dense networks composed of fine fibers [Citation19]. Patients with high fibrin density in their fibrin network have a higher risk of thrombotic events than patients with normal density [Citation20].
In patients with thrombotic CD, such as fibrinogen Paris V [Citation21] (FGAc.1717C>T) and fibrinogen Perth [Citation22] (FGAc.1541delC), fibrinolysis is prolonged. Fibrinogen Tokyo V was shown to have an amino acid substitution of γAla327Thr and extra glycosylation at γAsn325, accounting for the recurrent embolic episodes in the patient. In the crystal structure of the D region of fibrinogen, γAla327 resides near the calciumion binding site and the ‘a’ polymerization pocket. Thus, the substitution of γAla327Thr would likely interfere with calcium binding to this region and also affect the polymerization pocket. Furthermore, extra glycosylation of γAsn325 may interfere with the D:D association indirectly. Thromboembolism in these patients may result from fibrinolysis-resistant fragile clots and the formation of a large amount of soluble fibrin [Citation23]. Fibrinogen Melbourne (c.1055G>T, Cys326Phe) is a novel congenital hypodysfibrinogenemia caused by g326Cys-Phe in the fibrinogen γ chain, presenting as massive splanchnic venous thrombosis. The γ326Cys is located in the centre of an important t-TPA binding site, which possibly explains why the conformational change is associated with thrombosis [Citation24]. In γAla327Val, alanine is replaced by valine, affecting the D:D association, and hindering the formation of fibrils, thereby affecting the aggregation of fibrinogen.
The aggregation test and thromboelastography results in our proband and her family members revealed impaired fibrin aggregation. We did not find prolonged fibrinolysis or lysis-resistant clots in our patients with CD compared with healthy individuals, suggesting that the γAla327Val mutation does not affect fibrin clot dissolution.
In this study, in the proband, the concentration of fibrinogen activity was significantly lower than that of fibrinogen antigen (0.75 g/L vs. 1.59 g/L, reference range: 2–4 g/L). Immunoturbidimetry is based on the specific binding of fibrinogen antibody to fibrinogen. As fibrinogen antigen determinants are present in mutant fibrinogen molecules, the concentration of fibrinogen antigen in plasma of patients with CD will not be reduced. Combination of multiple methods to detect fibrinogen can reduce the rate of misdiagnosis and missed diagnosis of CD. Our previous study showed that the combination of a PT-derived method and the Clauss method is helpful in the screening for CD [Citation25]. Although no bleeding or thrombotic events have occurred in the family investigated in this study, the possibility that thrombotic or hemorrhagic events may occur in the future, especially in the case of surgery, delivery, or trauma, cannot be excluded. Alving et al [Citation26] reported a case of CD with an α Arg16His homozygous mutation (fibrinogen Giessen I), who had no bleeding or thrombotic events in daily life, but had symptoms of massive bleeding occurred during delivery. Therefore, the proband and her family still need follow-up to prevent thrombosis, bleeding, or other events.
Acknowledgements
We would like to thank the Department of Hematology and Clinical Laboratory of the First Affiliated Hospital of Guangxi Medical University, China. This research was supported by Grants from the National Natural Science Foundation of China (81560342).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Tennent GA, Brennan SO, Stangou AJ, et al. Human plasma fibrinogen is synthesized in the liver. Blood. 2007;109(5):1971–1974.
- Medved L, Weisel JW. Recommendations for nomenclature on fibrinogen and fibrin. J Thromb Haemost. 2010;7(2):355–359.
- Simurda T, Snahnicanova Z, Loderer D, et al. Fibrinogen martin: a novel mutation in FGB (Gln180Stop) causing congenital afibrinogenemia. Sem Thromb Hemost. 2016; 42(4):455–458.
- Yan J, Deng D, Luo M, et al. Dysfibrinogenemia in a patient undergoing artificial abortion after misdiagnosis and review of the literature. Clin Chim Acta. 2015;447:86–89.
- Luo M, Wei A, Xiang L, et al. Abnormal fibrinogen with an aalpha 16Arg –> Cys substitution is associated with multiple cerebral infarctions. J Thromb Thromb. 2018;46(3):409–419.
- Hamano A, Mimuro J, Aoshima M, et al. Thrombophilic dysfibrinogen Tokyo V with the amino acid substitution of gammaAla327Thr: formation of fragile but fibrinolysis-resistant fibrin clots and its relevance to arterial thromboembolism. Blood. 2004;103(8):3045–3050.
- Casini A, Neerman-Arbez M, Moerloose D P, et al. Dysfibrinogenemia: from molecular anomalies to clinical manifestations and management. J Thromb Haemost. 2015;13(6):909–919.
- Imurda T, Zolkova J, Snahnicanova Z, et al. Comparison of clinical phenotype with genetic and laboratory results in 31 patients with congenital dysfibrinogenemia in northern Slovakia. Int J Hematol. 2020;111:29.
- Brennan SO, Fellowes AP, Faed JM, et al. Hypofibrinogenemia in an individual with 2 coding (γ82 A→G and Bβ235 P→L) and 2 noncoding mutations. Blood. 2000;95(5):1709–1713.
- Neerman-Arbez M, de Moerloose P, Casini A. Laboratory and genetic investigation of mutations accounting for congenital fibrinogen disorders. Semin Thromb Hemost. 2016;42(4):356–365.
- Casini A, Blondon M, Tintillier V, et al. Mutational epidemiology of congenital fibrinogen disorders. Thromb Haemost. 2018;118(11):1867–1874.
- Everse SJ, Spraggon G, Veerapandian L, et al. Crystal structure of fragment double-D from human fibrin with two different bound ligands. Biochemistry. 1998;37(24):8637–8642.
- Weisel JW, Warren SG, Cohen C. Crystals of modified fibrinogen: size, shape and packing of molecules. J Mol Biol. 1978;126(2):159–183.
- Zhmurov A, Brown AE, Litvinov RI, et al. Mechanism of fibrin(ogen) forced unfolding. Structure. 2011;19(11):1615–1624.
- Ishikawa S, Hirota-Kawadobora M, Tozuka M, et al. Recombinant fibrinogen, gamma275Arg–>Cys, exhibits formation of disulfide bond with cysteine and severely impaired D:D interactions. J Thromb Haemost. 2004;2(3):468–475.
- Simurda T, Caccia S, Asselta R, et al. Congenital hypofibrinogenemia associated with a novel heterozygous nonsense mutation in the globular C-terminal domain of the γ-chain (p.Glu275Stop). J Thromb Thrombol. 2020;50(1):233–236.
- Cote HC, Lord ST, Pratt KP. Gamma-chain dysfibrinogenemias: molecular structure-function relationships of naturally occurring mutations in the gamma chain of human fibrinogen. Blood. 1998;92(7):2195–2212.
- Sugo T, Endo H, Matsuda M, et al. A classification of the fibrin network structures formed from the hereditary dysfibrinogens. J Thromb Haemost. 2006;4(8):1738–1746.
- Collet JP, Park D, Lesty C, et al. Influence of fibrin network conformation and fibrin fiber diameter on fibrinolysis speed: dynamic and structural approaches by confocal microscopy. Arterioscler Thromb Vasc Biol. 2000;20(5):1354–1361.
- Undas A, Ariens RA. Fibrin clot structure and function: a role in the pathophysiology of arterial and venous thromboembolic diseases. Arterioscler Thromb Vasc Biol. 2011;31(12):e88–e99.
- Collet JP, Soria J, Mirshahi M, et al. Dusart syndrome: a new concept of the relationship between fibrin clot architecture and fibrin clot degradability: hypofibrinolysis related to an abnormal clot structure. Blood. 1993;82(8):2462–2469.
- Westbury SK, Duval C, Philippou H, et al. Partial deletion of the alphaC-domain in the fibrinogen Perth variant is associated with thrombosis, increased clot strength and delayed fibrinolysis. Thromb Haemost. 2013;110(6):1135–1144.
- Hamano A, Mimuro J, Aoshima M, et al. Thrombophilic dysfibrinogen Tokyo V with the amino acid substitution of gammaAla327Thr: formation of fragile but fibrinolysis-resistant fibrin clots and its relevance to arterial thromboembolism. Blood. 2004;103(8):3045–3050.
- Cheah CY, Brennan SO, Kennedy H, et al. Fibrinogen Melbourne: a novel congenital hypodysfibrinogenemia caused by gamma326Cys-Phe in the fibrinogen gamma chain, presenting as massive splanchnic venous thrombosis. Blood Coagul Fibrinolysis. 2012;23(6):563–565.
- Xiang L, Luo M, Yan J, et al. Combined use of Clauss and prothrombin time-derived methods for determining fibrinogen concentrations: screening for congenital dysfibrinogenemia. J Clin Lab Anal. 2018;32(4):e22322.
- Alving BM, Henschen AH. Fibrinogen giessen I: a congenital homozygously expressed dysfibrinogenemia with A alpha 16 Arg-His substitution. Am J Hematol. 1987;25(4):479–482.