
ABSTRACT
Introduction
Liver iron overload is common in patients with thalassemia. In patients with beta-thalassemia, the correlation between serum ferritin and liver iron concentration is well established. The correlation between serum ferritin levels and liver iron concentrations in patients with alpha-thalassemia remains limited.
Methods
This is a cross-sectional study in patients with alpha-thalassemia aged ≥ 18 years old at Srinagarind Hospital, Khon Kaen University, Thailand. Liver iron concentration (LIC) was evaluated by the MRI-T2* technique. Linear logistic regression analysis was used to determine the correlation between serum ferritin levels and liver iron concentrations.
Results
One hundred and thirty-one of the MRI-T2* measurements from 65 patients with alpha-thalassemia were evaluated. Patients with non-deletional alpha-thalassemia had higher LIC compared to patients with deletional alpha-thalassemia. The serum ferritin levels were relatively low at the same levels of LIC in patients with non-deletional alpha-thalassemia compared to deletional alpha-thalassemia.
Conclusions
The correlation of serum ferritin levels and LIC was modest and different among alpha-thalassemia genotypes. A different serum ferritin threshold is needed to guide iron chelation therapy in patients with alpha-thalassemia. Evaluation of liver iron concentration is necessary for patients with alpha-thalassemia, especially in patients with non-deletional alpha-thalassemia.
Introduction
Non-transfusion-dependent thalassemia (NTDT) is a subgroup of thalassemic patients who have moderate disease severity and do not need lifelong regular red blood cell transfusion. NTDT consists of a wide variety of both beta-thalassemia and alpha-thalassemia genotypes, e.g. β-thalassemia intermedia, β-thalassemia/Hb E, and Hemoglobin H disease [Citation1].
Alpha-thalassemia (α-thalassemia) can be categorized as deletional α-thalassemia and non-deletional α-thalassemia. Deletional α-thalassemia is a genotype of thalassemia caused by deletional mutation of the α-globin genes leading to Hb H disease. Non-deletional α-thalassemia is a genotype of thalassemia caused by the non-deletion mutation of the α-globin genes, e.g. hemoglobin Constant Spring (Hb CS), and hemoglobin Paksé (Hb Paksé). Previous studies have demonstrated a heterogeneity of disease severity among α-thalassemia diseases. The non-deletional α-thalassemia diseases have more severe clinical symptoms in comparison to the deletional α-thalassemia diseases [Citation2,Citation3].
The pathophysiology of iron overload in NTDT caused by increased intestinal iron absorption due to ineffective erythropoiesis. Ineffective erythropoiesis leads to increase growth differentiation factor 15 (GDF15) levels resulted in suppression of the hepcidin levels [Citation4–6]. Erythroferrone (ERFE), one of the important hormones that regulate hepcidin levels. An increase in ERFE levels during stress erythropoiesis suppresses hepcidin levels [Citation7] may play a role in iron overload in thalassemia. Evaluation of ERFE, hepcidin levels, iron status, and erythroid activity would be beneficial for understanding ineffective erythropoiesis and iron overload in patients with alpha thalassemia.
Evidence has shown that serum ferritin levels had a good association with liver iron concentration (LIC) in patients with β-thalassemia disease [Citation8]. Taher et al. showed that the serum ferritin level that defined liver iron overload (LIC ≥ 5 mg Fe/g dw) was 800 ng/ml and with this threshold, the PPV and NPV were 91.7% and 53.6% in patients with NTDT. This study also demonstrated predictive values of serum ferritin threshold for guiding iron chelation therapy in these patients [Citation9]. In the results from this study, however, the majority of enrolled patients in this cohort were β-thalassemia intermedia, which may not apply in patients with α-thalassemia disease.
In patients with α-thalassemia, however, the association between LIC and serum ferritin levels remains limited. This study aimed to determine the association of LIC and serum ferritin in patients with α-thalassemia and identify the serum ferritin threshold for guiding iron chelation therapy in these patients.
Patients and methods
The study conducted in patients with α-thalassemia aged ≥ 18 years old at Srinagarind Hospital, Khon Kaen, Thailand. The medical histories and laboratory data of all recruited patients collected from the institutional thalassemia registry. Liver iron concentrations were evaluated by the MRI-T2* technique. The serum ferritin levels within 3 months of the MRI-T2* measurements were used in this cohort. Exclusion criteria were conditions that might cause high false serum ferritin levels such as the following: active infection /inflammation, cancer, or active hepatitis.
The research protocol was approved by the Ethics Committee of the Faculty of Medicine, Khon Kaen University (HE621342).
Statistical analyses
The correlation between LIC and serum ferritin levels demonstrated by the linear logistic regression analysis and reported as correlations r2. Predictive values (PPV and NPV) of LIC ≥ 3 mg Fe/g dw, LIC ≥ 7 mg Fe/g dw and the serum ferritin levels were calculated from the 2 × 2 table. The optimal cut-off point of the serum ferritin levels for predicting LIC ≥ 7 mg Fe/g dw derived from the receiver-operating characteristics curve. We used the STATA program version 10 (StataCorp, College Station, TX) to perform all statistical analyses. A p-value less than 0.05 was considered as a statistical significance.
Results
One hundred and thirty-one of the MRI-T2* measurements were from 65 patients with α-thalassemia (33 females, 32 males) were reviewed. Patients were divided into group A and group B. Group A was the patients with deletional α-thalassemia. Group B was those patients with non-deletional α-thalassemia. The mean age was higher in group A when compared with group B (35 years vs. 28 years). Mean serum ferritin levels were higher in patients with group A compared to those patients with group B (1008 ng/ml vs. 862 ng/ml), but mean LICs were comparable (8.2 mg Fe/g dw vs. 8.6 mg Fe/g dw). Splenectomy was more prevalent among patients with group B compared to those patients with group A (28% vs. 5%). Deferiprone (locally made in Thailand, GPOL1) is the first-line drug for oral iron chelation therapy in adult patients with thalassemia in Thailand. Therefore, all of the patients in both groups received deferiprone therapy during the study period. The most common genotype in group A was Hb H disease (12 patients, 54.5%) followed by the EABart’s disease (9 patients, 41%). In the group B, the most common genotype was compound heterozygous Hb H with Hb CS (20 patients, 46.5%), and the EABart’s disease with Hb CS (20 patients, 46.5%). Most of the patients in this cohort are non-transfusion-dependent thalassemia. None of the patients in group A is transfusion-dependent thalassemia. Five patients (11.6%) in group B are transfusion-dependent thalassemia. A summary of baseline clinical characteristics is shown in . There was a modest correlation between LIC and serum ferritin levels in patients with α-thalassemia (). The correlation was slightly lower in the group B (correlation r = 0.5, r2=0.3) compared to the group A (correlation r = 0.63, r2 = 0.4). shows a graph box relationship of the serum ferritin levels to the LIC ratios between the two groups. The group B patients had relatively lower serum ferritin levels in comparison to LIC.
Figure 1. Serum ferritin levels and liver iron concentrations among patients with alpha-thalassemia disease.
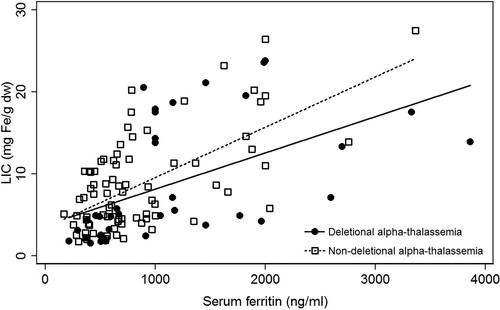
Figure 2. Grap box of serum ferritin to liver iron concentration ratios in patients with alpha-thalassemia disease.
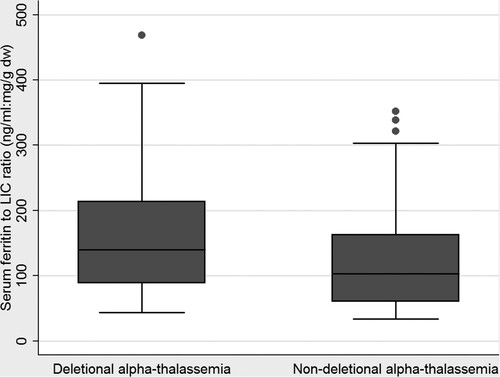
Table 1. Baseline clinical characteristics of 65 patients with alpha-thalassemia.
shows predictive values of serum ferritin levels for dose interruptions and dose escalation of iron chelation therapy. Using the cut-off point of serum ferritin < 300 ng/ml for dose interruptions according to the previous study in NTDT, the PPV and NPV were 89.8% and 84.3%. Using the cut-off point of serum ferritin > 2,000 ng/ml for dose escalation of iron chelation, the PPV and NPV were 28.3% and 100%. shows the ROC curve of serum ferritin levels for predicting moderate liver iron overload (LIC ≥ 7 mg Fe/g dw). The serum ferritin threshold of 600 ng/ml showed the highest sensitivity and specificity for predicting LIC ≥ 7 mg Fe/g dw (sensitivity 78.5% and specificity 59.1%) with an area under the ROC curve of 0.76 and 95% confidence intervals (CI) of 0.68–0.84.
Figure 3. The ROC curve of liver iron concentration ≥ 7 mg Fe/g dw and serum ferritin levels in patients with alpha-thalassemia disease.
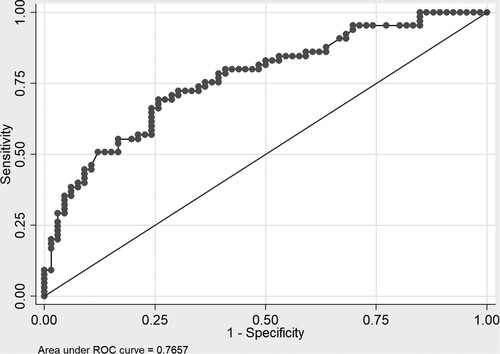
Table 2. Predictive values of serum ferritin of < 300 ng/ml for dose interruption and serum ferritin of > 2,000 ng/ml for dose escalation of iron chelation therapy in patients with alpha-thalassemia.
Discussion
The correlation between LIC and serum ferritin levels was modest in patients with α-thalassemia. The correlations were different among patients with non-deletional α-thalassemia and deletional α-thalassemia. The LIC was higher in patients with non-deletional α-thalassemia compared to those patients with deletional α-thalassemia at the same level of serum ferritin. These results may be explained by the β-thalassemia and α-thalassemia have differences in the erythropoiesis-hepcidin-iron axis. Origa R et al. showed that there were differences in the relationship between hepcidin levels, iron status, and erythroid activity among patients with thalassemia. The erythroid activity was significantly increased in patients with β-thalassemia intermedia leading to decreased hepcidin levels and increased serum ferritin levels [Citation10]. Previous studies showed a higher disease severity in patients with non-deletional α-thalassemia than patients with deletional Hb H disease [Citation2,Citation3]. Therefore, greater ineffective erythropoiesis in patients with non-deletional α-thalassemia (Hb CS and Hb Paksé in this cohort) may leaded to higher LIC and relatively low serum ferritin compared to deletional α-thalassemia. Supporting these results, the previous study in alpha-thalassemia also demonstrated differences in serum ferritin to LIC ratios among patients with α-thalassemia. They found that the serum ferritin to LIC ratio is lower in HbH with HbCS compared to Hb H disease [Citation11].
The previous study in patients with NTDT had demonstrated serum ferritin thresholds for guiding iron chelation therapy in patients with NTDT. Using the serum ferritin < 300 ng/ml for disruption of iron chelation according to the THALASSA study [Citation12,Citation13], good predictive values of PPV and NPV were 89.8% and 84.3% were shown in patients with α-thalassemia. Therefore, the serum ferritin < 300 ng/ml may be used as a cut-off level for interruption of iron chelation therapy in these patients. The serum ferritin > 2,000 ng/ml for increasing the dose of iron chelation, however, was not a good predictive value for LIC ≥ 7 mg Fe/g dw in patients with α-thalassemia. This result may be explained by the relatively low serum ferritin compared to the LIC in patients with α-thalassemia. In this cohort, the serum ferritin threshold of 600 ng/ml had the highest sensitivity and specificity for predicting LIC ≥ 7 mg Fe/g dw in patients with α-thalassemia.
These findings highlighted the need for liver iron concentration evaluation in patients with α-thalassemia, especially those patients with non-deletional α-thalassemia. A large study in patients with α-thalassemia is needed to find an appropriate serum ferritin threshold for guiding iron chelation treatment in these patients.
A limitation of this study is the small sample size of patients to demonstrate the appropriate serum ferritin threshold for guiding iron chelation treatment. Larger studies are needed to identify the appropriate serum ferritin levels for tailoring iron chelation therapy in patients with α-thalassemia. The different iron chelations may show different correlations between serum ferritin and liver iron concentrations. To the authors’ knowledge, this is the first and largest study of the correlation between LIC and serum ferritin levels in patients with α-thalassemia treated with deferiprone. The results from this study could be used as guidance to the management of iron overload in patients with alpha thalassemia.
In conclusion, the correlations between liver iron concentration and serum ferritin were modest and different among patients with α-thalassemia. Patients with non-deletional α- thalassemia had relatively low serum ferritin at the same level of LIC compared to patients with deletional α-thalassemia. Therefore, a different serum ferritin threshold is needed for guiding iron chelation therapy in patients with α-thalassemia. Evaluation of liver iron concentration is necessary for patients with α-thalassemia, particularly in those patients with non-deletional α-thalassemia.
Authors contributions
NT designed the study, collected data, performed statistical analysis, and wrote the manuscript; CS, KC, and AJ helped in data collection.
Acknowledgement
The authors would like to acknowledge Professor Vip Viprakasit, Professor Rungroj Krittayaphong, and Associate Professor Pairash Saiviroonporn (The Asia-Pacific network on iron overload assessment by MRI) for helping with the validation of the MRI-T2* measurements. The authors would like to thank Emeritus Professor James A. Will, University of Wisconsin–Madison, for helping in preparing the manuscript via publication clinic of Khon Kaen University, Thailand.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Weatherall DJ. The definition and epidemiology of non-transfusion-dependent thalassemia. Blood Rev. 2012;26(Suppl 1):S3–S6.
- Lal A, Goldrich ML, Haines DA, et al. Heterogeneity of hemoglobin H disease in childhood. N Engl J Med. 2011;364:710–718.
- Chen FE, Ooi C, Ha SY, et al. Genetic and clinical features of hemoglobin H disease in Chinese patients. N Engl J Med. 2000;343:544–550.
- Rivella S. The role of ineffective erythropoiesis in non-transfusion-dependent thalassemia. Blood Rev. 2012;26(Suppl 1):S12–S15.
- Musallam KM, Cappellini MD, Taher AT. Iron overload in β-thalassemia intermedia: an emerging concern. Curr Opin Hematol. 2013;20:187–192.
- Camaschella C, Nai A. Ineffective erythropoiesis and regulation of iron status in iron loading anaemias. Br J Haematol. 2016;172:512–523.
- Kautz L, Jung G, Valore EV, et al. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet. 2014;46:678–684.
- Taher A, El Rassi F, Isma’eel H, et al. Correlation of liver iron concentration determined by R2 magnetic resonance imaging with serum ferritin in patients with thalassemia intermedia. Haematologica. 2008;93:1584–1586.
- Taher AT, Porter JB, Viprakasit V, et al. Defining serum ferritin thresholds to predict clinically relevant liver iron concentrations for guiding deferasirox therapy when MRI is unavailable in patients with non-transfusion-dependent thalassaemia. Br J Haematol. 2015;168(2):284–290.
- Origa R, Cazzola M, Mereu E, et al. Differences in the erythropoiesis-hepcidin-iron store axis between hemoglobin H disease and β-thalassemia intermedia. Haematologica. 2015;100:e169–e171.
- Ang AL, Le TT, Tan RS. HbH constant spring disease has lower serum ferritin relative to liver iron concentration (LIC): importance of LIC measurement and potential impact on serum ferritin thresholds for iron chelation. Br J Haematol. 2017;176:986–988.
- Taher AT, Porter J, Viprakasit V, et al. Deferasirox reduces iron overload significantly in nontransfusion-dependent thalassemia: 1-year results from a prospective, randomized, double-blind, placebo-controlled study. Blood. 2012;120:970–977.
- Taher AT, Porter JB, Viprakasit V, et al. Deferasirox effectively reduces iron overload in non-transfusion-dependent thalassemia (NTDT) patients: 1-year extension results from the THALASSA study. Ann Hematol. 2013;92:1485–1493.