
ABSTRACT
Paroxysmal nocturnal hemoglobinuria (PNH) is a disease caused by a phosphatidylinositol glycan anchor biosynthesis class A (PIG-A) mutation in hematopoietic stem cells. There are three theories about the possible mechanism of the pathogenesis of PNH: immune escape, anti-apoptotic mechanism, and secondary gene mutation. There has been little gain in the knowledge regarding its pathogenesis during the last decade owing to the lack of representative cell lines and animal models. There have been recent reports about the successful creation of PNH mouse and PNH rhesus macaque models. The detection of glycosylphosphatidylinositol-anchor protein (GPI-AP)-deficient cells and/or fluorescently labeled variant of aerolysin (FLAER) test, estimation of erythrocyte life span, and hemolysis-related experiments demonstrated that these animal models of PNH had GPI-AP-deficient blood cells with shortened lifespans and increased sensitivity to complement-activated hemolysis. However, there were no clinical manifestations such as hemolysis and thrombosis in these animal models. This suggested that the PIG-A mutation is one of the several conditions required for PNH, but it alone is not enough to cause PNH.
Paroxysmal nocturnal hemoglobinuria (PNH) is caused by a PIG-A gene mutation in hematopoietic stem cells. The predominant clinical manifestations are hemolytic anemia, thrombosis, and bone marrow failure [Citation1]. PIG-A gene mutation on the X chromosome of hematopoietic stem cells leads to the dysfunction of glycosylphosphatidylinositol (GPI) synthesis and the deficiency of GPI anchor protein (GPI-AP) on the cell membrane [Citation2,Citation3]. Flow cytometry, the gold standard test for the clinical diagnosis of PNH, is used to detect GPI-APs such as CD55 and CD59, along with the application of FLAER test to detect GPI protein. Currently, the main treatments of PNH include controlling the onset of hemolysis, stimulating hematopoiesis, and preventing thrombosis. Recombinant human anti-complement 5 monoclonal antibodies have been used to significantly improve the prognosis of patients with PNH. However, since these drugs are not yet available in China, glucocorticoids remain the standard therapeutic agents. Glucocorticoids or monoclonal antibodies can reduce the complement-mediated lysis of PNH clones, thereby alleviating clinical symptoms such as hemolysis [Citation4,Citation5]. Elucidation of the pathogenesis of PNH has not advanced over the last decade, owing to the lack of representative cell lines and animal models. This limits the research and development of the targeted drugs. The relevant characteristics of the existing PNH animal models are summarized in this paper.
I. Progress in the pathogenesis of PNH
It is currently believed that PIG-A gene mutation is a prerequisite for the onset of PNH, and the proliferation of PNH clones is the necessary condition for the onset of PNH. However, PIG-A gene mutation alone is not sufficient to induce the proliferation of PNH clones [Citation6]. There are three theories about the possible mechanisms of clonal proliferation: immune escape, anti-apoptotic mechanism, and secondary gene mutation. The immune escape theory suggests that PNH clones lacking GPI-AP can escape the autoimmune surveillance normally mediated by T lymphocytes or may induce the immune system to selectively attack normal hematopoietic stem cells, thereby conferring clonal expansion advantages to PNH cells. The anti-apoptotic mechanism theory postulates, the PIG-A gene mutation itself induces inherent resistance to apoptosis, thereby giving PNH clones a proliferation advantage. The secondary mutation theory proposes that the non-neoplastic proliferation of hematopoietic stem cells in PNH patients may require the involvement of supplementary genetic defects, in addition to the PIG-A mutation.
Immune escape and anti-apoptotic mechanisms can explain the development and resistance of mutated PNH cells yet are not sufficient to describe the proliferation of PNH clones. Therefore, the possible effects of secondary mutated genes, other than PIG-A, have attracted much attention. Globally, researchers have used whole gene sequencing technology to screen for possible secondary mutated genes [Citation7]. Our research center conducted a full exon sequencing with 13 PNH patients and found multiple secondary mutated genes including RBPJ, MUC4, SUZ12, MAGEC1, CUX1, MLL2, TET2, and CEBP2 [Citation8]. Further in vitro tests revealed that the expression level of RBPJ was significantly higher in the CD59- than in CD59+ cells of PNH patients. When siRNA-RBPJ was used to inhibit the expression of RBPJ in PNH primary cells, their apoptosis rate increased, their proliferation activity decreased with the extension of transfection time, and the cells were blocked in the G0/G1 phase [Citation9]. However, due to the absence of adequate in vivo verification methods, the precise pathogenic gene and its mechanism of action has not been determined. This therefore necessitates the establishment of a PNH animal model to study the pathogenesis of the disease. Several domestic and foreign scholars have developed PNH animal models through PIG-A gene editing, but some typical clinical manifestations such as hemolysis have not been observed, thereby necessitating an improvement of the models. The successful establishment of a clinically relevant PNH animal model will be of great significance to explore the pathogenesis of the disease and identify new therapeutic targets.
II. Existing PNH animal models and their deficiencies
Gene knockout is a biotechnological method used to delete specific genes in animals. Conventional knockout (KO) and conditional knockout (CKO) can be achieved in animals. Currently, several animal models of PNH have been reported, including PNH mouse and PNH rhesus macaque models. In several varieties of PNH mouse models, gene KO was achieved by editing different loci of the Pig-A gene in mouse embryonic stem cells (ES cells). Pig-A gene knocked out within all cells and tissues of mice constitutes a complete KO mouse, while the Pig-A gene knocked out only in some cells and tissues of mice forms a CKO mouse.
1. Pig-A gene knockout mouse model
The human PIG-A gene is located on the X chromosome (Xp22.2), with a length of 17 kb. It encodes a subunit of the protein product 1-6N-acetylglucosamine transferase with 484 amino acids and is involved in the first step of GPI synthesis [Citation6]. Mutations of PIG-A lead to the failure of the normal synthesis of GPI protein and the loss of cell phenotype, which contributes to the pathogenesis of PNH [Citation10–12]. There are a variety of somatic PIG-A mutations in PNH patients, with more than a few hundred currently reported. Our research center sequenced genes from 33 PNH patients and found 26 mutants of the PIG-A gene, but without locating any hot spot mutations. Most mutations were single-base insertions or deletions, such as single or multiple base substitutions, while long sequence insertions or deletions were rare. Exon 2 was the region with the highest frequency of mutation [Citation13] and is also the most common editing region in gene KO animal models.
All PNH mouse models reported in literature adopted the ES cell target gene KO method [Citation14–20]. Gene targeting technology is a molecular biology method based on homologous DNA recombination within embryonic stem cells. Using homologous recombination technology, foreign genes are integrated into specific sites on the genome of the target cells, to modify chromosomes. The Pig-A gene of ES cells in mice were edited by the homologous recombination method. The target ES cells were screened and amplified, and then injected into the uterus of a surrogate mouse to form a chimeric inner cell group, and a non-functional gene chimeric animal was obtained [Citation21,Citation22].
1.1. Conventional Pig-A knockout mouse
In 1996, Kawagoe et al. [Citation14] first reported that editing exon 2 of the Pig-A gene using ES targeting techniques produced a conventional Pig-A KO mouse. The method resulted in a high rate of intrauterine fetal death of the chimeric mice, with a birth rate of only 10% and chimeric rate of less than 5% [Citation14]. This phenomenon may be related to the important function of the Pig-A gene in embryonic development. The proportion of GPI-AP-deficient red blood cells in mice at birth was found to correlate with the degree of chimerism in the KO mice [Citation14]. Owing to the low chimerism rate of KO mice, the proportion of blood cells with GPI-AP deficiency at birth, in the mouse model was low. Furthermore, the proportion of GPI-AP-deficient blood cells in KO mice reduced with the passage of postnatal time [Citation15], confirming that the Pig-A mutation alone was not sufficient to cause PNH clonal proliferation. Therefore, the KO mouse model of PNH cannot be used in PNH-related experimental studies because of the low birth rate and decreasing proportion of GPI-AP-deficient blood cells, with time.
1.2. Conditional Pig-A knockout mouse
The Cre/loxP system enables the knocking out of a target gene at a specific time or in a specific cell or tissue. Cre recombinase is derived from phage P1, and the sequence length is 38 bp. The LoxP sequence is about 34-bp long and consists of two 13-bp reverse repeats separated by an 8-bp non-palindromic sequence [Citation23]. Cre recombinase can act on two LoxP sites to cause specific homologous recombination and remove DNA sequences between LoxP sites located in the same direction [Citation24]. Since the Cre recombinant enzyme is controlled by the instantaneous expression promoter, which itself is only expressed and activated in specific cell types or tissues, it becomes possible to pinpoint a particular place to delete a DNA sequence and obtain a CKO mouse [Citation25].
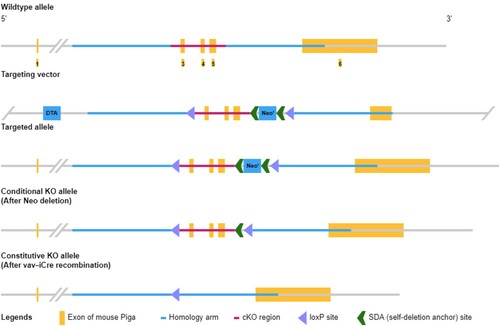
Pig-A CKO mice were first constructed by inserting a LoxP site in the same direction at each end of the Pig-A gene sequence, and then the Flox mice were mated with the Cre recombinase mouse, so that the Pig-A sequence inserted in LoxP was deleted in the specific cell type or tissue expressing Cre. Pig-A was knocked out in certain cells by mating with different Cre mice. Pig-A CKO mice had a higher chimerism rate and GPI-AP-deficiency ratio, plus the proportion of GPI-AP-deficient blood cells was also more stable [Citation16,Citation17]. Keller et al. [Citation19] used Fes-Cre to induce Pig-A knockout in pluripotent hematopoietic stem cells, and the resulting mouse model showed Pig-A deletion in all hematopoietic cells and a stable ratio of GPI-AP-deletion blood cells. Jasinski et al. [Citation20] directed the knockout of the Pig-A gene in red blood cells using GATA1-Cre. The GPI-AP deletion ratio in red blood cells of the KO mice after birth could reach nearly 100% and the ratio was stable. Pig-A CKO mice have stable GPI-AP-deficient blood cells and can thus serve as animal models of PNH for disease-related studies. By using Vav-iCre, our institution successfully obtained Pig-A CKO mice whose Pig-A gene in hematopoietic stem cells was knocked out (). The expression of GPI protein and GPI-AP in all blood cells was absent, and the ratio was stable in the model (Supplementary Figure 1).
Figure 1. Model diagram of Pig-A gene knockout in a mouse subjected to gene editing by the Cre/loxP system in our center.
2. PIG-A gene knockout rhesus macaque model
Tae-hoon Shin successfully constructed PNH rhesus macaque models of PIG-A gene KO in hematopoietic stem cells through CRISPR/Cas9 gene editing [Citation26]. The CRISPR/Cas9 system is made up of the CRISPR gene and the Cas9 endonuclease. CRISPR is a DNA fragment that contains short repeats (20–50 bp) [Citation27], which are found in more than 40 percent of bacterial and 90 percent of archaeal genomes. The Cas gene (CRISPR-related gene) encodes a nuclease and helicase protein that unwinds and cuts DNA sequences. The CRISPR/Cas9 system repairs location-specific DNA double-strand breaks caused by Cas9-targeted chromosome cutting through non-homologous end joining (NHEJ), and then inserts abnormal bases or causes base loss at the fracture site, resulting in frameshift mutations, to finally achieve the knockout of the target [Citation28]. Using CRISPR/Cas9 to edit, PIG-A genes of rhesus macaque hematopoietic stem cells were deleted, and the edited cells were injected back into the animal to obtain the PNH rhesus macaque model.
3. Evaluation and existing problems of PNH animal models
The evaluation parameters for PNH mouse models include blood routine examination, flow cytometry, erythrocyte life span, and hemolytic tests. The results of blood routine examination of PNH mice reported in the literature show no anemia or elevated reticulocyte levels [Citation15,Citation16,Citation19]. Jasinski suggested that this was due to the late timing of lox-PigA gene recombination, which made red blood cells in PNH mouse models more similar to type II PNH cells, where hemolysis and anemia may only be mild, if not absent [Citation20]. Kawagoe speculated that GPI-AP might have a different role in regulating hematopoiesis in humans compared to mice [Citation14]. Murakami et al. suggested that this might be due to lower complement activity in mice, especially in C57BL/6 mice, and the red blood cells of mice have transmembrane complement regulatory molecules, such as complement receptor 1-related gene/protein y (Crry) [Citation29] and transmembrane accelerated aging factor (DAF) [Citation30]. PNH red blood cells in people with complete DAF/ CD59 deficiency are 10–15 times more sensitive to complement than normal red blood cells [Citation31]. However, the sensitivity of mouse GPI-AP-deficient red blood cells to complement action was only 3 times that of the normal red blood cells, and the presence of transmembrane complement regulating molecules on mouse red blood cells was the main reason for this difference [Citation16].
In addition to blood routine examination, PNH mice could also be evaluated through the percentage of GPI-AP-deficient cells. This is the most direct and simplest method for the diagnosis of PNH. The sequence of hematopoietic cells involved in human PNH cloning is granulocyte → monocyte → erythrocyte → lymphocyte, and to establish the diagnosis of PNH, at least two GPI anchor proteins of one or more types of hematopoietic cells should be missing [Citation32,Citation33]. In a mouse model, the number of GPI-AP-deficient blood cells can be identified by detecting the thermostable antigen (CD24/HSAg) on the surface of mature red blood cells and granulocytes, and CD48 on the surface of lymphocytes. CKO mice had a slightly higher and more stable percentage of GPI-AP-deficient blood cells than KO mice [Citation16,Citation17], and the rate of deletions did not change over time, after birth. This again proved that PIG-A mutation is one of the necessary conditions for PNH pathogenesis, yet PIG-A mutation alone is not enough to cause PNH clonal proliferation [Citation34,Citation35]. In addition, flow cytometric detection of GPI-AP on different cell surfaces in mouse models showed that abnormally cloned PNH cells had higher deletion ratios and stability in lymphocytes than other cell types [Citation15]. This was not identified in clinical patients, and the possible specific underlying mechanism is not clear.
In the PNH rhesus macaque model, FLAER was used to represent the number of GPI deficient blood cells. FLAER acts on all GPI anchors and does not cause errors due to the different types and amounts of GPI-AP expressed by different cells. Therefore, this detection method for GPI anchors was more sensitive and specific for diagnosing PNH [Citation36]. FLAER detection of granulocytes, monocytes, and lymphocytes in the PNH rhesus macaque model, permitted the dynamic monitoring of changes in PNH cloning in rhesus macaques. The results showed that abnormal PNH clones in rhesus macaques equally had no proliferation advantage [Citation26], consistent with results from the PNH mouse model.
It is well known that PNH patients have significantly shortened erythrocyte lifespans. Tremml et al. [Citation17] investigated the half-life of GPI-AP-deficient red blood cells in PNH mice and found it to be 7.3 (5.3–11.9) days, compared to 15 days for normal mice [Citation37]. He suggested that increased complement sensitivity may be one of the reasons for the reduced erythrocyte lifespan. Other possible mechanisms cannot be ruled out, such as macrophages in the reticuloendothelial system, which may also be involved in the clearance of red blood cells lacking GPI-AP. Meanwhile, through an acidifying serum hemolysis test performed on PNH mouse cells in-vitro, it was observed that the complement-mediated erythrocytic sensitivity of Pig-A cells increased, although the actual source mice did not have hemoglobinuria and obvious signs of hemolysis [Citation17]. The existing animal models of PNH are summarized in .
Table 1. Summary of existing PNH animal models.
III. Outlook
Although the molecular defects underlying PNH have been clarified, its pathogenesis, especially the mechanism of abnormal clonal proliferation, is still not fully understood. The animal models of PNH constructed by PIG-A gene editing did not show the typical clinical manifestations and blood alterations characteristic of the disease. However, the detection of GPI-AP-deficient cells and/or FLAER, erythrocyte life span estimation, and hemolysis experiments proved that the animal models of PNH had GPI-AP-deficient blood cells with a shortened lifespan and increased sensitivity to complement-activated hemolysis.
PIG-A mutation is one of the necessary conditions for PNH, but PIG-A mutation alone is not enough to cause PNH clonal proliferation. Researchers have confirmed through gene sequencing methods that secondary gene mutations other than PIG-A and their resulting expressions may be involved in the abnormal proliferation of PNH clones. In the future, it is hoped that our institution can further verify the influence of the secondary gene mutation on the proliferation of PNH clones using the constructed PNH animal models. The animal models of PNH are of great significance to further study the pathogenesis of this disease and to find new therapeutic targets.
Supplemental Material
Download TIFF Image (526.3 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Related Research Data
References
- Rotoli B, Luzzatto L. Paroxysmal nocturnal hemoglobinuria. Semin Hematol. 1989, Jul;26(3):201–207.
- Bessler M, Mason PJ, Hillmen P, et al. Paroxysmal nocturnal haemoglobinuria (PNH) is caused by somatic mutations in the PIG-A gene. EMBO J. 1994;13(1):110–117.
- Ware RE, Rosse WF, Howard TA. Mutations within the Piga gene in patients with paroxysmal nocturnal hemoglobinuria. Blood. 1994;83(9):2418–2422.
- Ra B. How I treat paroxysmal nocturnal hemoglobinuria. Blood. 2009;113:6522–6527.
- Rong F. How do I treat paroxysmal nocturnal hemoglobinuria. Chin J Hematol. 2008;39(11):887–891.
- Liyan L, Rong F. Pathogenesis of paroxysmal nocturnal hemoglobinuria. Chin J Hematol. 2008;39(6):527–528.
- Shen W, Clemente MJ, Hosono N, et al. Deep sequencing reveals stepwise mutation acquisition in paroxysmal nocturnal hemoglobinuria. J Clin Invest. 2014;124(10):4529–4538.
- Li LY, Liu ZY, Liu H, et al. Deep sequencing of whole genome exon in paroxysmal nocturnal hemoglobinuria. Am J Hematol. 2017 Apr;4(92):E51–E53.
- Li L, Liu H, Wang H, et al. Abnormal expression and mutation of the RBPJ gene may be involved in CD59-clonal proliferation in paroxysmal nocturnal hemoglobinuria. Exp Ther Med. 2019 Jun;17(6):4536–4546.
- Brodsky RA, Mukhina GL, Nelson KL, et al. Resistance of paroxysmal nocturnal hemoglobinuria cells to the gly-cosylphosphatidylinositol-binding toxin aerolysin. Blood. 1999;93(5):1749–1756.
- Fujita M, Kinoshita T. GPI-anchor remodeling: potential functions of GPI-anchors in intracellular trafficking and membrane dynamics. Biochim Biophys Acta. 2012 Aug;1821(8):1050–1058.
- Kinoshita T, Fujita M, Maeda Y. Biosynthesis, remodelling and functions of mammalian Gpi-anchored proteins: recent progress. J Biochem. 2008;144(3):287–294.
- Park J, Kim M, Kim Y, et al. Clonal cell proliferation in paroxysmal nocturnal hemoglobinuria: evaluation of PIGA mutations and T-cell receptor clonality. Ann Lab Med. 2019;39(5):438–446.
- Kawagoe K, Kitamura D, Okabe M, et al. Glycosylphosphatidylinositol-anchor-deficient mice: implications for clonal dominance of mutant cells in paroxysmal nocturnal hemoglobinuria. Blood. 1996;87(9):3600–3606.
- Rosti V, Tremml G, Soares V, et al. Murine embryonic stem cells without pig-a gene activity are competent for hematopoiesis with the PNH phenotype but not for clonal expansion. J Clin Invest. 1997;100(5):1028–1036.
- Murakami Y, Kinoshita T, Maeda Y, et al. Different roles of glycosylphosphatidylinositol in various hematopoietic cells as revealed by a mouse model of paroxysmal nocturnal hemoglobinuria. Blood. 1999;94(9):2963–2970.
- Tremml G, Dominguez C, Rosti V, et al. Increased sensitivity to complement and a decreased red blood cell life span in mice mosaic for a nonfunctional Piga gene. Blood. 1999;94(9):2945–2954.
- Takahama Y, Ohishi K, Tokoro Y, et al. Functional competence of T cells in the absence of glycosyl phosphatidylinositol-anchored proteins caused by T cell-specific disruption of the Pig-agene. Eur J Immunol. 1998;28(7):2159–2166.
- Keller P, Payne JL, Tremml G, et al. Fes-Cre targets phosphatidylinositol glycan class a (Piga) inactivation to hematopoietic stem cells in the bone marrow. J Exp Med. 2001;194(5):581–589.
- Jasinski M, Keller P, Fujiwara Y, et al. GATA1-Cre mediates Piga gene inactivation in the erythroid/megakaryocytic lineage and leads to circulating red cells with a partial deficiency in glycosyl phosphatidylinositol-linked proteins (paroxysmal nocturnal hemoglobinuria type II cells). Blood. 2001;98(7):2248–2255.
- Capecchi MR. The new mouse genetics: altering the genome by gene targeting. Trends Genet. 1989;5(3):70–76.
- Galli-Taliadoros LA, Sedgwick JD, Wood SA, et al. Gene knock-out technology: a methodological overview for the interested novice. J Immunol Methods. 1995;181(1):1–15.
- Kühn R, Torres RM. Cre/loxP recombination system and gene targeting. Methods Mol Biol. 2002;180:175–204.
- Wang CH, Sun H. Progress in gene knockout mice. Sheng Wu Gong Cheng Xue Bao. 2019;35(5):784–794 (in Chinese).
- Kawano F, Okazaki R, Yazawa M, et al. A photoactivatable Cre-LoXP recombination system for optogenetic genome engineering. Nat Chem Biol. 2016;12(12):1059–1064.
- Shin TH, Baek EJ, Corat MAF, et al. CRISPR/cas9 PIG-A gene editing in nonhuman primate model demonstrates no intrinsic clonal expansion of PNH HSPCs. Blood. 2019;133(23):2542–2545.
- Mojica FJ, Ferrer C, Juez G, et al. Long stretches of short tandem repeats are present in the largest replicons of the archaea haloferax mediterranei and Haloferax volcanii and could be involved in replicon partitioning. Mol Microbiol. 1995;17(1):85–93.
- Hsu PD, Lander ES, Zhang F. Development and applications of Crispr-Cas9 for genome engineering. Cell. 2014;157(6):1262–1278.
- Molina H, Wong W, Kinoshita T, et al. Distinct receptor and regulatory properties of recombinant mouse 1 complement receptor (CR1) and Crry, the two genetic homologues of human CR1. J Exp Med. 1992;175(1):121–129.
- Spicer AP, Seldin MF, Gendler SJ. Molecular cloning and chromosomal localization of the mouse decay-accelerating factor genes. Duplicated genes encode glycosylphosphatidylinositol-anchored and transmembrane forms. J Immunol. 1995;155(6):3079–3091.
- Rosse WF, Adams JP, Thorpe AM. The population of cells in paroxysmal nocturnal haemoglobinuria of intermediate sensitivity to complement lysis: significance and mechanism of increased immune lysis. Br J Haematol. 1974;28(2):181–190.
- Manivannan P, Ahuja A, Pati HP. Diagnosis of paroxysmal nocturnal hemoglobinuria: recent advances. Indian J Hematol Blood Transfus. 2017;33(4):453–462.
- Keeney M, Illingworth A, Sutherland DR. Paroxysmal nocturnal hemoglobinuria assessment by flow cytometric analysis. Clin Lab Med. 2017;37(4):855–867.
- Brodsky RA. How do pig-a mutant paroxysmal nocturnal hemoglobinuria stem cells achieve dominance. Expert Rev Hematol. 2009;2(4):353–356.
- Hill A, DeZern AE, Kinoshita T, et al. Paroxysmal nocturnal haemoglobinuria. Nat Rev Dis Primers. 2017;3(3):17028.
- Dahmani A, Roudot H, Cymbalista F, et al. Evaluation of fluorescently labeled aerolysin as a new kind of reagent for flow cytometry tests:optimization of use of FLAER, Hints, and Limits. Am J Clin Pathol. 2016;145(3):407–417.
- Russell ES, Bernstein SE. Blood and blood formation. In: Green EL, editor. Biology of the laboratory mouse. New York: McGraw-Hill Book Company ; 1966. p. P351–372.