
ABSTRACT
Background
Among myeloproliferative neoplasms, it is often difficult to distinguish essential thrombocythaemia (ET) from prefibrotic-stage primary myelofibrosis (PMF) with thrombocytosis given their overlapping clinicopathological phenotypes.
Case presentation
We encountered a 45-year-old male who was initially diagnosed with ET and eventually became transformed to secondary myelofibrosis 20 years later. Two distinct types of aberrant megakaryocytes were observed at diagnosis: one type characteristic of ET and the other type characteristic of PMF. With a proliferation in the bone marrow, aberrant megakaryocytes were infiltrated into the extramedullary organs and were even present in the thrombus were observed at autopsy. As a result of next-generation sequencing, the significant increase of variant allele frequency (VAF) of JAK2 V617F and U2AF1 S34Y mutations was observed in the bone marrow cells at the final stage.
Conclusions
This patient could be recognized as an atypical case of aggressive megakaryocytosis transformed from ET.
Introduction
Patients diagnosed with one of the myeloproliferative neoplasms (MPNs) show heterogeneous clinicopathological phenotypes. Essential thrombocythaemia (ET) and primary myelofibrosis (PMF) constitute two distinct entities of three BCR-ABL1-negative MPNs. It is often difficult to distinguish patients with ET from those with prefibrotic stage PMF with thrombocytosis. Here, we report a 45-year-old male who was initially diagnosed with ET, and the condition transformed to secondary myelofibrosis (MF) 20 years later. At diagnosis, his megakaryocytes were morphologically divided into two groups: some characteristic of ET and the others characteristic of prefibrotic MF. Unexpectedly, at the final stage of his clinical course, dysplastic megakaryocytes proliferated in the bone marrow and infiltrated into the extramedullary organs. As a result of next-generation sequencing at this stage, the significant increase of variant allele frequency (VAF) of U2AF1 S34Y mutation was observed coupled with an increase of JAK2 V617F mutation in the bone marrow cells.
Case presentation
A 45-year-old male experienced dizziness while driving and was admitted to a local hospital. An elevated haemoglobin level was noted on two recent consecutive annual medical check-ups. His blood analysis revealed pancytosis (WBC 23,100 /μl, RBC 577 × 104 /μl, Hb 18.1 g/dl, Plt 68.7 × 104 /μl), and he was referred to our institute. On physical examination, an enlarged spleen was palpable 2 fingerbreadths below the left costal margin. No other clinical symptom related to dysregulated proinflammatory cytokines or disturbed microvascular circulation or splenomegaly was noted. Laboratory findings in our institute also revealed pancytosis: WBC 19,600/μl (Stab 5.5%, Seg 74.5%, Eosino 2.0%, Baso 1.0%, Mono 3.5%, Lymph 13.5%), RBC 592 × 104 /μl, Hb 18.2 g/dl, and Plt 66.6 × 104 /μl (). The white blood cell differential count exhibited slight dominancy of segmented neutrophils without erythroblasts in the peripheral blood. Serum erythropoietin (EPO) level (13.8 mU/ml), circulating red blood cell volume (24.6 ml/kg) and partial pressure of oxygen in arterial blood (PaO2) (87.7 mmHg) were all within normal range (). An initial bone marrow aspiration revealed hypercellularity (nucleated cell count (NCC): 51.5 × 104/μl) with an increased number of megakaryocytes (900/μl). Percentage of erythroid precursors (erythroblast: Ebl) accounted for 21.2% of total nucleated cells. The myeloid to erythroid (M:E) ratio was slightly elevated (3.3) (). A needle biopsy of the bone marrow performed after an aspiration revealed slightly hyperplastic bone marrow accompanied by an increased number of dysplastic megakaryocytes (a, b). The megakaryocytes exhibited two distinct morphological types. One type had abundant and mature cytoplasm and deeply lobulated or hyperlobulated nuclei often referred to as 'staghorn-like', whereas the other type had an altered nuclear:cytoplasmic ratio with abnormal chromatin clumping and plump (cloud-like) or bulbous lobulation of the nuclei (balloon-shaped) [Citation1]. Both types of dysplastic megakaryocytes formed loose clusters. Silver staining of the bone marrow biopsy specimen revealed a very loose network of reticulin fibre that was categorized as grade 0 myelofibrosis (MF-0) according to the criteria of the European consensus on grading bone marrow fibrosis (c, d) [Citation2]. Chromosomal analysis of the peripheral blood revealed a normal male karyotype, and the BCR-ABL1 fusion gene was negative based on a fluorescent in situ hybridization analysis. We could not seek for the presence of JAK2 mutation at that time. Collectively, we diagnosed this patient as ET but not PV, considering that serum EPO level and circulating red blood cell volume were normal. Cytoreductive therapy with nimustine was commenced and followed by administration of hydroxyurea 15 months later ().
Figure 1. Bone marrow histology in low-power (a and c; x100) or high-power (b and d ∼ h; x400) fields at diagnosis (a ∼ d), transformation into fibrosis (e and g) and the exaggerated thrombocytosis phase (f and h). a, b: H-E staining revealed slightly hyperplastic bone marrow with an increased number of dysplastic megakaryocytes. Two distinct types of dysplastic megakaryocytes were observed. One type exhibited abundant and mature cytoplasm and deeply and hyperlobulated nuclei (staghorn-like), whereas the other type exhibited an altered nuclear:cytoplasmic ratio with abnormal chromatin clumping and plump (cloud-like) or bulbous (balloon-shaped) lobulation of the nuclei. These two types of abnormal megakaryocytes formed loose clusters. Arrows and arrowheads indicate abnormal megakaryocytes with staghorn-like and cloud-like nuclei, respectively. c, d: Silver staining of the initial bone marrow biopsy revealed a very loose network of reticulin fibre, and the condition was categorized as grade 0 myelofibrosis (MF-0) based on the criteria of the European consensus on grading bone marrow fibrosis. e, f: H-E staining revealed infiltration of dysplastic megakaryocytes in the secondary (e) and tertiary (f) analyses. g, h: Silver staining of the secondary (g) and tertiary (h) bone marrow biopsy revealed obvious fibrosis. Compared with the secondary biopsy (g), the tertiary sample (h) exhibited more prominent fibrosis.
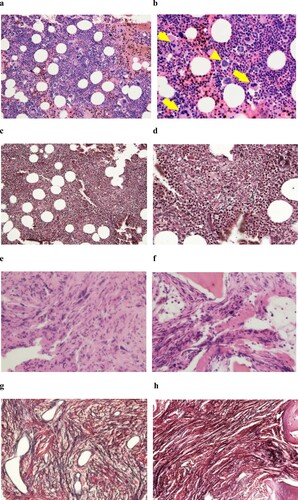
Figure 2. Clinical course.
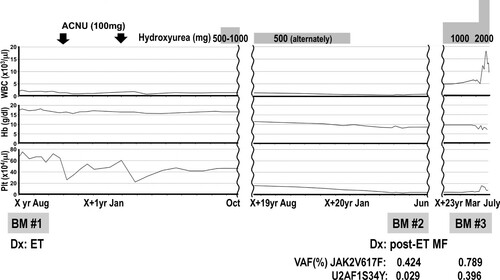
Table 1. Laboratory findings.
Approximately 20 years after the initial diagnosis, anaemia and thrombocytopenia continuously progressed. The platelet count continued to decrease even after the discontinuation of hydroxyurea. The haemoglobin level also decreased to 9.2 g/dl from the baseline (18.2 g/dl), which was a decrease by more than 2 g/dl, and obvious leukoerythroblastosis was noted. LDH levels exceeded the reference level. Under the suspicion of fibrotic transformation, we reanalysed the bone marrow. All these findings met the criteria of fibrotic transformation in the WHO criteria. Unlike the initial study, the bone marrow was successfully aspirated (e). Coarse bundles of collagen fibres with dense reticulin fibres were observed in silver staining of the trephine biopsy specimen (g), and grade 3 myelofibrosis (MF-3) was confirmed based on the European consensus on grading bone marrow fibrosis. With these consecutive data, we verified myelofibrotic transformation under the criteria for post-ET MF [Citation3]. In addition, the presence of a JAK2 V617F heterozygous mutation was confirmed by using quantitative PCR analysis at this time ().
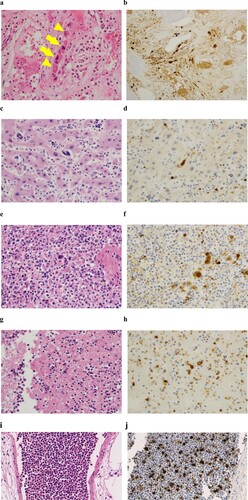
Approximately 3 years after the discontinuation of hydroxyurea, leukocyte counts began to increase. The third bone marrow examination was performed one month after readministration of hydroxyurea and revealed more extensive myelofibrosis compared with the former examination (f, h). Unfortunately, despite the administration of an escalated dose of hydroxyurea, several clinical symptoms possibly related to dysregulated proinflammatory cytokines, such as fever, general malaise and night sweats, emerged along with exaggerated leukocytosis. Even after being admitted, these lines of symptoms continued to progress, and the patient died 6 days after hospitalization. We did not have the opportunity to administer ruxolitinib. An autopsy revealed marked hepatosplenomegaly. The liver and spleen weighted 4,000 g and 2,000 g, respectively. White and red thrombi of various sizes were observed in the vena cava inferior and bilateral pulmonary arteries. The latter might have caused lethal haemodynamic deterioration. Along with extensive fibrosis, the proliferation of both types of dysplastic megakaryocytes characteristic of ET and PMF remained in the bone marrow (a). The abnormal megakaryocytes extensively infiltrated in non-haematopoietic organs, including the liver and spleen (c, e), and were detected even in arterial and venous thrombi, and cell aggregate in pulmonary blood vessels (g, i). These megakaryocytes were positive for CD42b (b, d, f, h, j) but negative for CD34.
Figure 3. H-E staining of the bone marrow (a), liver (c), spleen (e), thrombus (g) and lung (i) at autopsy. Infiltration of dysplastic megakaryocytes was confirmed with anti-CD42b immunostaining in the bone marrow (b), liver (d), spleen (f), thrombus (h) and lung (j). Arrows and arrowheads indicate abnormal megakaryocytes with staghorn-like and cloud-like nuclei, respectively (a).
In order to clarify the molecular basis on distinct changes of clinical features in the terminal stage, the secondary (at the diagnosis of post-ET MF) and tertiary (at the further progression of myelofibrosis) bone marrow samples were subjected to next-generation sequencing analysis [Citation4]. As a result, a significant increase of VAFs of both JAK2 V617F and U2AF1 S34Y mutations was observed in the bone marrow cells (0.424% to 0.789%, and 0.029% to 0.396%, respectively) (). There was no mutation of CALR or MPL genes in either sample.
Discussion
Up to 30% of PMF patients are asymptomatic but are consistently associated with thrombocytosis [Citation1]. It is difficult to make an accurate differential diagnosis between ET and early PMF with thrombocytosis. In addition, 30 - 40% of PMF patients exhibit prefibrotic or early fibrotic stage disease [Citation1]. In both stages, an increased number of megakaryocytes is the exclusive positive finding without obvious fibrosis in the bone marrow [Citation1]. To address this issue, PMF is further subclassified into two stages in the World Health Organization (WHO) classification of myeloid neoplasms revised in 2017 (WHO 2017 classification): prefibrotic/early and overt fibrotic stages [Citation5]. In this revision, the significance of morphological findings is emphasized to differentiate ET patients from PMF patients in the prefibrotic stage (prePMF). The most striking abnormalities of megakaryocytes in ET patients include abundant and mature cytoplasm and deeply lobulateded and hypersegmented (staghorn-like) nuclei. In contrast, the important morphological findings in PMF patients include deviations from the normal nuclear:cytoplasmic ratio suggestive of defective maturation; abnormal patterns of chromatin clumping exhibiting hyperchromatic, cloud-like, or balloon-shaped nuclei; and bare nuclei [Citation5]. Based on the WHO 2017 classification, the patient’s clinical manifestation and laboratory data did not fulfil the criteria for ET or prePMF at the time of administration. Histopathological analysis of the bone marrow samples, especially megakaryocytes, should have been critical to distinguish these two types of diseases. This patient’s bone marrow exhibited increased cellularity compared with the typical bone marrow of ET patients. No obvious dominance of erythroid precursor cells was noted. Intriguingly, two distinct types of dysplastic megakaryocytes were concurrently observed: one with ‘staghorn-like’ nuclei typically observed in ET and the other with ‘cloud-like’ or ‘balloon-shaped’ nuclei in PMF. Both types of dysplastic megakaryocytes were increased in number and formed loose clusters.
The discriminatory power of the WHO classification criteria remains controversial, especially between ET and prefibrotic/early PMF [Citation6]. Various factors, such as disease diversity and interobserver discordance, may affect this problem [Citation6, Citation7]. To address this issue, a multicentre study with a total of 295 patients with MPNs was performed, focusing on validation of the discriminatory power of the WHO classification system [Citation8]. As a result, acceptable consensus concerning the discrimination between ET and PMF was achieved in 88% of cases. However, among the MPN patients analysed in this study, a few cases could not be categorized into any typical disease entities. Some peculiar patients exhibited mixed characters of polycythaemia vera (PV) and ET and were categorized as ‘ET/PV’. The other undistinguishable patients were categorized as MPN-U. It is quite interesting that no patient exhibited a mixed appearance of ET and PMF similar to that noted in this patient even in this large study. Therefore, this patient is rare and valuable for solving the discriminatory issue between these two difficult-to-distinguish diseases. It was difficult to obtain an accurate diagnosis in this patient at the time of initial administration even under the newly revised WHO 2017 classification.
Patients with PMF generally exhibit significantly worse clinical outcomes compared with ET patients. The median overall survival of patients with ET and prefibrotic PMF from initial diagnosis is 21 and 14 years, respectively [Citation8]. The ten-year and 15-year survival rates of ET and early/prefibrotic PMF are 89% and 76% and are 80% and 59%, respectively [Citation9]. Myelofibrotic transformation along with the development of acute myeloid leukaemia in MPN patients is a potentially fatal complication, and the condition is evident earlier in early/prefibrotic MF compared with ET. The frequency of myelofibrotic transformation in ET patients is 0.8-4.9% and 4-11% at 10 and 15 years after diagnosis, respectively [Citation10], whereas the frequency in early/prefibrotic PMF patients is 2.3%, 12.3% and 16.9% at 5, 10 and 15 years, respectively [Citation9]. In this patient, apparent bone marrow fibrosis was detected more than 20 years after diagnosis. Although the initial findings were inconsistent with the typical pathologic manifestations of ET, our patient collectively exhibited an ET clinical course rather than PMF. Moreover, given that the median time to fibrotic transformation in ET patients is approximately 7–16 years from the time of diagnosis [Citation10], transformation was delayed in this patient compared with the average duration. Seven risk factors, including advanced age (≧ 60), leukocytosis (≧ 11,000/μl), anaemia, reticulin fibrosis and increased cellularity of the bone marrow, absence of the JAK2 V617F mutation, use of anagrelide and the presence of an ASXL1 mutation, affect fibrotic transformation in ET patients [Citation10]. Although the mutational status of JAK2 or ASXL1 was not tested, the clinical findings of our patient met only two of the seven factors, namely, leukocytosis (19,600 /μl) and hypercellularity of the bone marrow at the time of diagnosis, which could have prolonged the period before the transformation.
In ET patients, a homozygous JAK2 V617F mutation is rare (2.2%) and is associated with stimulated erythropoiesis, myelopoiesis, reduced platelet count, increased incidence of splenomegaly, increased spleen size, an increased necessity of cytoreductive therapy, an increased risk of thrombotic events and fibrotic transformation [Citation11]. Approximately 3 years after confirming JAK2 V617F heterozygosity (VAF, 0.424), the VAF was increased to 0.789, indicating that the mutation became homozygous at least in some part of the cell population. In parallel, fibrotic transformation became more prominent, and the patient died of thrombosis in the pulmonary arteries. This clinical course is essentially consistent with previous reports regarding clinical correlates of the JAK2 V617F allele burden in MPNs [Citation12].
The most outstanding pathological feature of this patient at autopsy was extensive infiltration of dysplastic megakaryocytes in the extramedullary lesions. In ET patients, leukaemic transformation is quite rare [Citation13]. Dysplastic megakaryocytes, but not megakaryoblasts, proliferated in the bone marrow of this patient. Dysplastic megakaryocytes were also observed not only in the representative organs for pathological extramedullary haematopoiesis, i.e. the spleen and liver, but also in thrombi and in cell aggregate in blood vessels of the lung. This finding might represent evidence that pathological megakaryocytes circulated in the peripheral blood. Recently it is reported that megakaryocytes migrate out of the bone marrow to the lungs, where they produce platelets accounting for 50% of total platelet production [Citation14]. In this patient, however, there was no evidence that thrombopoiesis in the lung was established. Actually, the number of platelets was below the level of normal range. CD34 is typically expressed on leukaemic blasts transformed from MPNs. An anti-CD34 antibody failed to detect megakaryocytes in the bone marrow of this patient. This patient could be recognized as exhibiting an aberrant type of megakaryocytosis transformed from ET.
As a result of next-generation sequencing analysis, a significant increase of VAFs of both JAK2 V617F and U2AF1 S34Y mutations was observed when compared between secondary (at the diagnosis of post-ET MF) and tertiary (at the further progression of myelofibrosis) bone marrow samples (0.424% to 0.789%, and 0.029% to 0.396%, respectively) (). U2AF1 gene encodes RNA splicing factor, and its mutations (S34Y and S34F) were firstly identified in the bone marrow cells derived from patients of myelodysplastic syndromes (MDS) [Citation15], and play a significant role in leukemogenesis in myeloid malignancies [Citation16]. U2AF1 mutations are also observed in patients with PMF at a frequency of 16%. [Citation17, Citation18]. Although the underlying molecular mechanism is not fully elucidated, U2AF1 mutations are significantly correlated with anemia and thrombocytopenia, and are recognized as adverse factors associated with inferior overall or leukemia-free survival [Citation17]. In addition, co-occurrence of mutation of MPN-associated driver genes (JAK2, CALR or MPL) and spliceosome genes is observed in certain myeloid malignancies [Citation19], such as JAK2 V617F and SF3B1 mutation in MDS/MPN with ring sideroblasts and thrombosis (MDS/MPN-RS-T) [Citation20, Citation21]. These lines of evidence imply that the U2AF1 mutation also may have contributed to an aggressive proliferation of mature megakaryocytes at the final stage in this case.
In summary, considering the clinicopathological features of this patient that megakaryocytes of ET and prefibrotic MF types were mixed up in the bone marrow, it might have been impossible to clearly discriminate ET and prefibrotic MF at diagnosis and predict the clinical course. The occurrence of U2AF1 S34Y mutation at least partly could have modified the clinical course and accelerated fibrosis in the bone marrow. However, the molecular basis on the rapid production of mature infiltrative megakaryocytes at the final stage remains unknown. It is interesting to analyse how mutations of MPM drivers and spliceosome genes collaborate molecularly. We need further examination on clinical information from a large number of patients to clarify whether their co-existence determines some specific entities in MPN.
Acknowledgements
We give special thanks to Dr. Soji Morishita and Prof. Norio Komatsu in the Department of Haematology, Juntendo University Graduate School of Medicine for precise mutational analysis of MPL and CALR.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Swerdlow SH, Campo E, Harris NL, et al. WHO classification of tumors of haematopoietic and lymphoid tissues. Lyon: IARC press, 2008. p. 40–50.
- Thiele J, Kvasnicka HM, Facchetti F, et al. European consensus on grading bone marrow fibrosis and assessment Of cellularity. Haematologica. 2005;90:1128–1132.
- Barosi G, Mesa RA, Thiele J, et al.; international working group for myelofibrosis research and treatment (IWG-MRT). proposed criteria for the diagnosis of post-PV & post-ET MF; a consensus statement from the international working group for myelofibrosis research and treatment. Leukemia. 2008;22:437–438.
- Ochi Y, Kon A, Sakata T, et al.; combined cohesin-RUNX1 deficiency synergistically perturbs chromatin looping and causes myelodysplastic syndromes. Cancer Discov. 2020;10(6):836–853.
- Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–2405.
- Barbui T, Thiele J, Vannucchi AM, et al. Problems and pitfalls regarding WHO-defined diagnosis of early/prefibrotic primary myelofibrosis versus essential thrombocythemia. Leukemia. 2013;27:1953–1958.
- Wilkins BS, Erber WN, Bareford D, et al. Bone marrow pathology in essential thrombocythemia: interobserver reliability and utility for identifying disease subtypes. Blood. 2008;111:60–70.
- Thiele J, Kvasnicka HM, Müllauer L, et al. Essential thrombocythemia versus early primary myelofibrosis: a multicenter study to validate the WHO classification. Blood. 2011;117:5710–5718.
- Barbui T, Thiele J, Passamonti F, et al. Survival and disease progression in essential thrombocythemia are significantly influenced by accurate morphologic diagnosis: an international study. J Clin Oncol. 2011;29:3179–3184.
- Cerquozzi S, Tefferi A. Blast transformation and fibrotic progression in polycythemia vera and essential thrombocythemia: a literature review of incidence and risk factors. Blood Cancer J. 2015;5:e366.
- Vannucchi AM, Antonioli E, Guglielmelli P, et al. Clinical profile of homozygous JAK2 617V > F mutation in patients with polycythemia vera or essential thrombocythemia. Blood. 2007;110:840–846.
- Vannucchi AM, Pieri L, Guglielmelli P. JAK2 allele burden in the myeloproliferative neoplasms: effects on phenotype, prognosis and change with treatment. Ther Adv Hematol. 2011;2:21–32.
- Radaelli F, Mazza R, Curioni E, et al. Acute megakaryocytic leukemia in essential thrombocythemia: an unusual evolution? Eur J Haematol. 2002;69:108–111.
- Lefrançais E, Ortiz-Muñoz G, Caudrillier A, et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature. 2017;544:105–109.
- Graubert T A, Shen D, Ding L, et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Letter) Nature Genet. 2012;44:53–57.
- Przychodzen B, Jerez A, Guinta K, et al. Patterns of missplicing due to somatic U2AF1 mutations in myeloid neoplasms. Blood. 2013;122(6):999–1006.
- Tefferi A, Lasho TL, Finke CM, et al. Targeted deep sequencing in primary myelofibrosis. Blood Adv. 2016;1(2):105–111.
- Tefferi A, Finke CM, Lasho TL, et al. U2AF1 mutation types in primary myelofibrosis: phenotypic and prognostic distinctions. Leukemia. 2018;32(10):2274–2278.
- Liu YC, Illar GM, Bailey NG. Clinicopathologic characterisation of myeloid neoplasms with concurrent spliceosome mutations and myeloproliferative-neoplasm-associated mutations. J Clin Pathol. 2020;73(11):728–736.
- Patnaik MM, Lasho TL, Finke CM, et al. Predictors of survival in refractory anemia with ring sideroblasts and thrombocytosis (RARS-T) and the role of next-generation sequencing. Am J Hematol. 2016;91:492–498.
- Jeromin S, Haferlach T, Weissmann S, et al. Refractory anemia with ring sideroblasts and marked thrombocytosis cases harbor mutations in SF3B1 or other spliceosome genes accompanied by JAK2V617F and ASXL1 mutations. Haematologica. 2015;100:e125–e127.