
ABSTRACT
Over the past 20 years, granulocyte colony-stimulating factor (G-CSF) has driven the attention of researchers as a therapeutic agent for curing patients suffering from neutropenia. Despite the successful use of G-CSF, it currently requires daily injections, which are inconvenient, expensive, and distressing for children. Therefore, an alternative strategy for using G-CSF for treatment is needed. Understanding the G-CSF structure, expression, mechanism of action, and how it induces neutrophils mobilization is crucial to producing promising cancer therapy. The ability of G-CSF to mobilize hematopoietic stem cells from the bone marrow into the blood circulation was consequently exploited and altered the practice of hematopoietic stem cell transplantation. This is the motivation for the current review, which sheds light on the history of G-CSF and then focuses on the mechanism of action upon binding to its receptor (G-CSFR) and how that had led to the stimulation of neutrophils mobilization. The findings of this review show new insight into the mechanism of G-CSF that induces neutrophils mobilization. Thus, Understanding the G-CSF will provide a more effective treatment for all neutropenia patients.
KEYWORDS:
1. History of granulocyte colony-stimulating factor (G-CSF)
In the early 1960s, several studies on animal models were conducted to explore how white blood cells (WBCs) are regulated within the blood circulation. In 1966, the identification of WBCs specific regulator remained unknown until two research groups developed an in-vitro assay that measured the growing colonies of granulocytes and monocytes from bone marrow (BM) and spleen cells samples [Citation1,Citation2]. Nevertheless, the growth of colonies was based on the presence of unknown proteins that were given the name of colony-stimulating factors (CSFs). In the middle of the 1980s, different laboratories performed work to purify and classify CSF proteins. Resulting from these efforts, four CSF proteins with different activities were discovered. They were classified and named based on the type of cell colonies they stimulated: granulocyte-macrophage colony stimulating factor (GM-CSF) stimulated both granulocyte and macrophage colonies, macrophage colony stimulating factor (M-CSF) stimulated macrophage colonies, G-CSF stimulated granulocyte colony formation, and multi-CSF (known as interleukin 3, IL-3) stimulated multiple hematopoietic cell colonies [Citation3].
In the middle of 1983, G-CSF was first purified and characterized in mice using a mouse lung-conditioned medium by Nicola and his collaborators in Melbourne, Australia [Citation4]. After two years, Human GCSF (hG-CSF) was first purified from the human bladder carcinoma cell line 5637 [Citation5]. In 1986, molecular cloning of the complementary deoxyribonucleic acid (cDNA) for G-CSF and the first expression of Escherichia coli (E. coli) were attained by Souza and Boone [Citation6]. As a result, the previous accomplishments facilitated the development of recombinant G-CSF, which enabled the study of its biological characteristics [Citation7].
2. G-CSF
2.1. Structure of G-CSF
Human G-CSF (hG-CSF) is located on chromosome 17 and encoded by CSF3 gene. However, this gene encodes two different messenger ribonucleic acid (mRNA) products due to G-CSF differential splicing: G-CSFa contains 177 amino acids (18.8kD) and G-CSFb contains 174 amino acids (19.6kD). The difference between the two types is that G-CSFa contains additional three residues after Leucine35 (Valine-Serine-Glycine). The G-CSFb (174 amino acids) contains a glycosylation site on the oxygen known as O- linked glycosylation. It is attached to one threonine at the site of residue 133, and this form is expressed in mammalian cells. It has been reported that G-CSFb obtains more biological activity (∼20 times more than G-CSFa), which makes it the source of commercial pharmaceutical products for G-CSF [Citation8].
The feature of hG-CSF central structure is similar to other helical cytokine family members. It contains four antiparallel, left-handed α-helical fold bundles connected by two long loops in a form that two helices extend up (A contains 29 amino acids & B contains 21 amino acids) and two helices extend down (C contains 24 amino acids & D contains 30 amino acids) [Citation9] (). Additionally, hG-CSF has five cysteine residues. Four of these cysteines form two internal disulfide bonds located between Cys36– Cys42 and Cys64– Cys74, leaving one free cysteine residue at Cys17th position with a free sulfhydryl group [Citation10].
Figure 1. Human G-CSF structure.
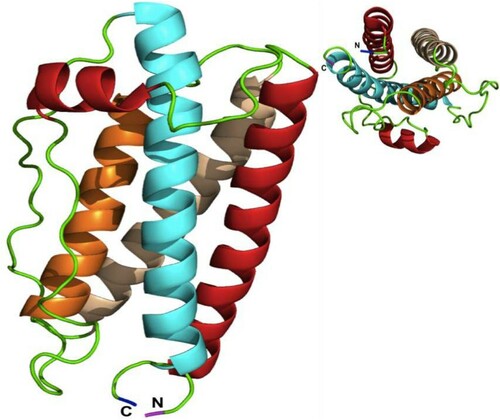
The central structure of hG-CSF consists of four antiparallel, left-handed α-helical fold bundles connected by two long loops in a form called up–up–down-down structure that two helices A (Red color) and B (Orange color) extend up and two helices C (White color) and D (Cyne color) extending down. N = amine-terminus and C = carboxyl-terminus (Molecular graphics system called PyMOL) ().
2.2. The importance of G-CSF expression and action
hG-CSF is a cytokine that regulates the proliferation, differentiation, and survival of neutrophils [Citation11], as proved by the significant reduction of neutrophils in both G-CSF and G-CSFR deficient mice [Citation12,Citation13]. G-CSF can be produced by a variety of cells, including endothelial cells, fibroblasts, macrophages, monocytes, and bone marrow stromal cells in response to several inflammatory mediators such as vascular endothelial growth factor (VEGF), interleukin β (IL-1β), interleukin (IL-17), lipopolysaccharide (LPS), necrosis factor alpha (TNF-α) [Citation14]. It has been reported that hG-CSF is highly expressed on a number of cancer cell types, including human gastric and colon cancers [Citation15,Citation16], as well as acute myeloid leukemia (AML) [Citation17,Citation18] and other different carcinoma cells [Citation19]. Besides, it has a major role in treating neutropenia in cancer patients undergoing chemotherapy [Citation20].
G-CSF is present at low levels in healthy individuals and at increased levels in infections and inflammations, etc [Citation21–23]. Normally, levels of circulating G-CSF are very low (<100 pg/mL). Nevertheless, G-CSF levels can increase to 20 times baseline levels in conditions of stress, resulting in a rapid increase of circulating neutrophils [Citation24].
Many mechanisms or evidences revealed that G-CSF could stimulate and regulate the production of neutrophils progenitors from the bone marrow to the blood circulation. G-CSF enhances the proliferation of all granulocytic lineages from myeloblast (hematopoietic stem cell) to myelocyte. GCSF drives differentiation of neutrophils and rapidly accelerates the metamyelocytes maturation, resulting in rapid and continuous elevation of neutrophils number in the blood circulation [Citation25–28]
Significantly, it has been shown that G-CSF treatments stimulate a faster erythropoiesis-enhancing response than that of erythropoietin (EPO). These data recommend an alternative method to treat acute anemia, especially when patients undergo a clinical emergency in remote areas without appropriate supplies from blood banks [Citation29].
Recently, it has been reported that G-CSF has a dual activity that is beneficial both in decreasing acute neuronal degeneration and enhancing long-term plasticity following cerebral ischemia in the CNS. Treatment of G-CSF exerts neuroprotective effects on damaged neurons throughout the suppression of the mitochondrial stress and endoplasmic reticulum (ER) stress and maintains cellular homeostasis by reducing pro-apoptotic proteins and increasing anti-apoptotic proteins [Citation30].
3. G-CSF receptor
3.1 Discovery, expression, and cloning of G-CSF receptor
Effects of G-CSF are mediated by binding to a single homodimer receptor, granulocyte colony stimulating factor receptor (G-CSFR). Therefore, the regulation, proliferation, and differentiation of neutrophils precursors are highly dependent upon binding to their receptors [Citation15]. GCSF-R is a membrane protein expressed in all granulocytic lineage cells, including neutrophils, progenitors, and myeloid leukemia cells [Citation31]. G-CSFRs have also been detected on normal B & T lymphocytes, monocytes [Citation32,Citation33], and non-hematopoietic tissues, such as cardiomyocytes [Citation34], vascular endothelial cells [Citation35], neural stem cells [Citation36], placenta [Citation37], and many non-hematopoietic tumors cell lines [Citation38].G-CSFRs are mainly expressed on common myeloid precursors (CMP), and mature neutrophils, however, the expression of these receptors increases more during maturation [Citation39]. Recently, G-CSFRs have been shown to be highly expressed on human gastric and colon cancer cells [Citation16].
In 1990, G-CSFR was first cloned from mice myeloid leukemia cell line (NFS-60) and shown to form homo-dimers upon binding to its ligand G-CSF, resulting in a complex 2:2 ligands: receptor subunit [Citation40].
3.2. G-CSFR structure and function
The G-CSFR is a cell surface receptor and belongs to the class I hematopoietic cytokine receptor super-family (HCR) [Citation41]. G-CSFR is around 120 kDa and contains 813 amino acids in length, arranged as follows: an extracellular region (604 amino acids), a transmembrane region (26 amino acids), and 183 amino acids for an intracellular cytoplasmic domain. The extracellular domain consists of N-terminal immunoglobulin (Ig)-like domain, cytokine receptor homology (CRH) domain, and a Trp-Ser-X-Trp-Ser (WSXWS) motif required for G-CSF ligand binding (a hallmark of the class I cytokine receptors), and the remainder of this region is formed by 3 fibronectin type III (FNIII) domains [Citation42–44].
The intracellular region contains three conserved sub-domains called Box 1, Box 2 and Box 3 [Citation45]. Box 1, Box 2, and tyrosine residues at site Y704 have a fundamental role in proliferating the signaling, while the Box 3 motif is associated with receptor trafficking [Citation46,Citation47]. The intracellular region also has three tyrosine residues, 729, 744, and 764, which play an important role in proliferation, differentiation, and cell survival () [Citation43,Citation46].
Figure 2. The scheme proposed for domains and downstream signal pathways of G-CSFR.
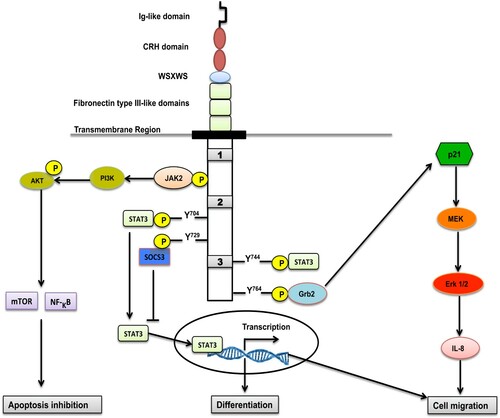
Ligation of G-CSF to its receptor forms homo-dimers with stoichiometry 2:2 [Citation48,Citation49]. Each ligand of G-CSF interacts with the CRH domain of one G-CSFR subunit and the immunoglobulin (Ig)-like domain of the second G-CSFR subunit, forming a crossover configuration of the receptor subunits ((B)) [Citation50].
Figure 3. [A] In the absence of the ligand, G-CSFR is associated with Janus kinases (Jaks). [B] The binding of the ligand to the receptor occurs at a 2:2 ligand:receptor subunit stoichiometry, forming a crossover configuration between the receptor subunits that brings the Jaks into proximity and enables their trans-phosphorylation and stimulation. [C] The intracellular 4-tyrosine residues of the G-CSFR (represented by stars) are phosphorylated by Jaks. [D] STAT interacts with the phosphotyrosine residues through their Src Homology 2 (SH2) domains and become phosphorylated by the Jak. Phospho-dimers of STATs accumulate in the nucleus and activate transcription factors that drive the neutrophils from the bone marrow to the blood circulation.
![Figure 3. [A] In the absence of the ligand, G-CSFR is associated with Janus kinases (Jaks). [B] The binding of the ligand to the receptor occurs at a 2:2 ligand:receptor subunit stoichiometry, forming a crossover configuration between the receptor subunits that brings the Jaks into proximity and enables their trans-phosphorylation and stimulation. [C] The intracellular 4-tyrosine residues of the G-CSFR (represented by stars) are phosphorylated by Jaks. [D] STAT interacts with the phosphotyrosine residues through their Src Homology 2 (SH2) domains and become phosphorylated by the Jak. Phospho-dimers of STATs accumulate in the nucleus and activate transcription factors that drive the neutrophils from the bone marrow to the blood circulation.](/cms/asset/7a6bd761-ec42-4c46-9ae8-83b187d0e832/yhem_a_1965725_f0003_oc.jpg)
3.2.1. JAK/STAT pathway
The binding of G-CSF causes conformational changes to its receptor that activates the Janus kinase (JAKs)/signal transducer family JAK1, JAK2, and Non-receptor tyrosine-protein kinase (TYK2) [Citation46]. The activation of JAKs proteins subsequently phosphorylates G-CSFR by binding to its Box 1 and 2 domains, creating potential docking sites for a variety of signaling molecules such as signal transducer and activator of transcription (STAT) proteins in cytoplasm [Citation51], particularly STAT3 with slight stimulation of STAT1 and STAT5 [Citation43,Citation52]. G-CSFR dimerizes and brings the JAKs together into proximity resulting in their trans-phosphorylation of one another. This, in turn, phosphorylates tyrosine (Y) residues (Y704, Y729, Y744, and Y764) located in the cytoplasmic region and serves as docking sites for STAT's. Notably, STAT3s have been reported to interact with tyrosine residues 704 and 744 of the G-CSFR through their Src Homology 2 (SH2) domains, get phosphorylated and activated by JAK2, and then form homodimers that migrate to the nucleus, where they bind DNA and activate gene transcription [Citation53–58]. It seems that they might regulate the mobilization of neutrophils from the bone marrow to the blood circulation [Citation59,Citation60]. In normal conditions, STAT3 induces a suppressor of cytokine signaling 3 (SOCS3) to bind and activate G-CSFR, leading to receptor degradation and cessation of the signalling [Citation58,Citation61]. SOCS proteins, mainly SOCS3, showed an inhibitory effect on G-CSF signaling during neutrophilic differentiation [Citation59–61].
STAT5 is another important signalling activated and mediated by G-CSFR that induces proliferation and survival of neutrophils [Citation62], directly controlled and activated by JAKs throughout phosphorylation [Citation61]. However, activation kinetics for STAT5 is significantly different from STAT3. The activation of STAT5 is transient and returns to the basal level in around half-hour, while the activation of STAT3 continues for many hours [Citation42,Citation61]. Therefore, binding of G-CSF to its receptor-mediated differential activation of both STAT5 and STAT3 may play an important role in controlling myeloid lineage proliferation against differentiation, especially neutrophils [Citation58]. Since activation of STAT3 is sustained for many hours, it could be the main regulator for the neutrophil's mobilization.
3.2.2. MAPK/ERK pathway
Although ligation of G-CSF to G-CSFR is widely believed to induce JAK/STAT pathways, it has also been linked to mitogen-activated protein kinases/extracellular signal-regulated protein kinase MAPK/ERK pathway (also recognized as the Ras-Raf-MEK-ERK pathway). Trosine residue Y764 serves as a docking site for the growth factor receptor-bound protein 2(Grb2), which induces p21 Ras pathway. In vitro, a significant decrease in the activation of p21 Ras and proliferation of neutrophils were noticed when Y764 was absent [Citation63,Citation64]. (Erk 1/2) MAP kinase is considered to be the main downstream effector from the p21 Ras pathway that is involved in signaling proliferation of myeloid precursor cells. It is also reported that Erk1/2 is strongly activated in neural cells upon exposure to G-CSF [Citation7,Citation42,Citation65]. The binding of G-CSF to its receptor activates the intracellular kinase (MEK). Phosphorylated MEK stimulates Erk ½ to be activated. Phosphorylated Erk ½ induces neutrophils migration and IL-8 production [Citation55].
3.2.3. AKT/PI3-K
AKT/ PI3-K signalling is another pathway that is activated by JAK2. It is important for differentiation, proliferation, and survival of immature neutrophil precursors by activating the nuclear factor kappa B/ mammalian target of rapamycin (NF-κB, mTOR), which acts as an inhibitor for apoptosis but is not able to extend the lifespan of neutrophils in the presence of G-CSF () [Citation66].
Mobilization of neutrophils
Production, proliferation, and differentiation of neutrophils initiate from the HSCs in the bone marrow. It has been shown that following differentiation; the majority of mature neutrophils remain in the bone marrow for approximately 5–6 days. During infection or inflammation, these cells are available for rapid response and release to the blood circulation and then migrate to affected tissues [Citation67].
G-CSF is a hematopoietic cytokine that regulates granulopoiesis and promotes proliferation, differentiation, and neutrophil activation [Citation14]. It has been shown that G-CSF enhances neutrophils’ migration into the peripheral tissues. In addition to G-CSF, different agents are involved in the regulation of neutrophils migration. For example, C5a, leukotriene B4 (LTB4), and CXCR2 ligands (e.g. IL-8) are found in humans, whereas keratinocyte chemoattractant [KC] and macrophage inflammatory protein 2 [MIP-2] in mice; [Citation68–70]. It has been shown that CXCR2 is more potent than G-CSF in enhancing neutrophils migration [Citation71].
Previously, it was reported that STAT3 is the major transcription factor activated upon binding of G-CSF to its receptor, but the role of STAT3 in the mechanism of neutrophils mobilization was not clear [Citation72]. At the early stage of acute inflammation, the chemokines, macrophage inflammatory protein-2 (MIP-2, known as Cxcl2) and keratinocyte-derived chemokine (KC, Cxcl1), and their shared receptor CXCR2 induce the mobilization of neutrophils from the BM to the circulating blood. A previous study examined the role of STAT3 for neutrophil migration by treating STAT3-deficient mice with MIP-2 by intraperitoneal injection. Treated STAT3-deficient mice did not show an increase in circulating neutrophil amounts compared to wild-type mice, which suggested the importance of STAT3 in neutrophils migration [Citation59,Citation60,Citation73].
Bajrami et al. [Citation67,Citation74] showed that G-CSF does not synergize with CXCR2 to induce neutrophil mobilization during the early phase of acute inflammation. Instead, it inhibits CXCR2-mediated rapid neutrophil mobilization. This result verifies that the initial CXCR2-mediated neutrophil mobilization can occur at maximal levels without G-CSF–induced inhibition. At a later-phase of acute inflammation, G-CSF could induce STAT3 to suppress CXCR2-mediated rapid neutrophil mobilization and reserved neutrophils in the BM, in part throughout their chemokine receptor 4 (CXCR4) expression, which binds to the stromal cell-derived-1 (SDF-1), expressed in the BM [Citation59,Citation60,Citation73] ().
Figure 4. Scheme of the proposed model. How G-CSF and STAT3 induce the mobilization of neutrophils. At the early stage of acute inflammation, MIP-2, KC, and their shared receptor CXCR2 induce the mobilization of neutrophils from the BM to the blood circulation. At the late stage of acute inflammation, GCSF, together with STAT3, inhibits CXCR2-mediated rapid neutrophil mobilization and reserved neutrophils in the BM, in part throughout their CXCR4 expression, which binds to SDF-1 (stromal cells express SDF-1) expressed in the BM. (↓ reserve ؛ ↑ induce).
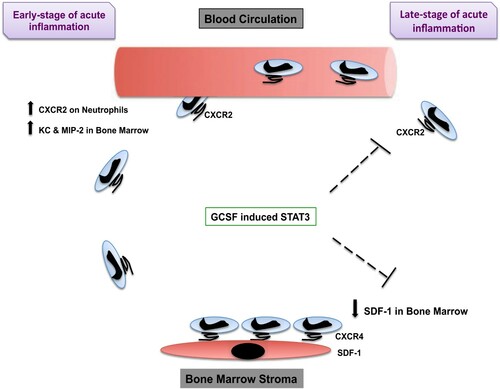
In summary, because G-CSF itself is not chemotactic, this concept is supported by the observation that G-CSF fails to induce circulating neutrophil amounts in CXCR2-knockout mice [Citation74]. Inhibiting the SDF-1/CXCR4 interaction is sufficient to enable neutrophil release from the marginated pool present in the lung and block the neutrophil trafficking back to the BM, as shown by the use of the CXCR4 antagonist AMD3100 (Plerixafor) [Citation75].
Conclusions
G-CSF is a hormone produced by different tissues to stimulate neutrophils’ production from the bone marrow into the blood circulation. The rhG-CSF has been shown to stimulate neutrophils to treat neutropenic patients and in stem cell mobilization in the cases of BM transplantation. Nowadays, G-CSF is a recognized therapy routinely used to treat patients with neutropenia and thus decreasing morbidity, especially cancer patients undergoing chemotherapeutic drug treatments. Understanding the G-CSF structure, expression, and mechanism of action upon binding to its receptor and leading to the mobilization of neutrophils is fundamental to generating a promising cancer therapy.
Declaration
Ethics approval and consent to participate
This study doesn't contain any human materials so there is no need for consent.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflict of interest.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Bradley TR, Metcalf D. The growth of mouse bone marrow cells in vitro. Aust J Exp Biol Med Sci. 1966;44(3):287–299.
- Ichikawa Y, Pluznik DH, Sachs L. In vitro control of the development of macrophage and granulocyte colonies. Proc Natl Acad Sci U S A. 1966;56(2):488–495.
- Metcalf D. The colony-stimulating factors and cancer. Nat Rev Cancer. 2010;10(6):425–434.
- Nicola NA, Metcalf D, Matsumoto M, et al. Purification of a factor inducing differentiation in murine myelomonocytic leukemia cells. identification as granulocyte colony-stimulating factor. J Biol Chem. 1983;258(14):9017–9023.
- Welte K, Platzer E, Lu L, et al. Purification and biochemical characterization of human pluripotent hematopoietic colony-stimulating factor. Proc Natl Acad Sci U S A. 1985;82(5):1526–1530.
- Souza LM, Boone TC, Gabrilove J, et al. Recombinant human granulocyte colony-stimulating factor: effects on normal and leukemic myeloid cells. Science. 1986;232(4746):61–65.
- Panopoulos AD, Watowich SS. Granulocyte colony-stimulating factor: molecular mechanisms of action during steady state and ‘emergency’ hematopoiesis. Cytokine. 2008;42(3):277–288.
- Aapro MS, Bohlius J, Cameron DA, et al. Update of EORTC guidelines for the use of granulocyte-colony stimulating factor to reduce the incidence of chemotherapy-induced febrile neutropenia in adult patients with lymphoproliferative disorders and solid tumours. Eur J Cancer. 2010;47(1):8–32.
- Arvedson T, Giffin M. Structural Biology of G-CSF and Its Receptor, in Twenty Years of G-CSF, G. Molineux, M. Foote, and T. Arvedson, Editors. 2012, Springer Basel. p. 61-82.
- Werner JM, Breeze AL, Kara B, et al. Secondary structure and backbone dynamics of human granulocyte colony-stimulating factor in solution. Biochemistry. 1994;33(23):7184–7192.
- Cox GN, et al. Hematopoietic properties of granulocyte colony-stimulating factor/immunoglobulin (G-CSF/IgG-Fc) fusion proteins in normal and neutropenic rodents. PLoS One. 2014;9(3):1–8.
- Lieschke GJ, Grail D, Hodgson G, et al. Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood. 1994;84(6):1737–1746.
- Liu F, Wu HY, Wesselschmidt R, et al. Impaired production and increased apoptosis of neutrophils in granulocyte colony-stimulating factor receptor-deficient mice. Immunity. 1996;5(5):491–501.
- Gregory AD, Hogue LA, Ferkol TW. Regulation of systemic and local neutrophil responses by G-CSF during pulmonary pseudomonas aeruginosa infection. Blood. 2007;109(8):3235–3243.
- Gascon P. Presently available biosimilars in hematology-oncology: G-CSF. Target Oncol. 2012;7(1):29–34.
- Morris KT, Khan H, Ahmad A, et al. G-CSF and G-CSFR are highly expressed in human gastric and colon cancers and promote carcinoma cell proliferation and migration. Br J Cancer. 2014;110(5):1211–1220.
- El Fakih R, Rasheed W, Hawsawi Y, et al. Targeting FLT3 mutations in acute myeloid leukemia. Cells. 2018;7(1):1–9.
- Kotb A, El Fakih R, Hanbali A, et al. Philadelphia-like acute lymphoblastic leukemia: diagnostic dilemma and management perspectives. Exp Hematol. 2018;67:1–9.
- Beekman R, Valkhof MG, Sanders MA, et al. Sequential gain of mutations in severe congenital neutropenia progressing to acute myeloid leukemia. Blood. 2012;119(22):5071–5077.
- Mehta HM, Malandra M, Corey SJ. G-CSF and GM-CSF in neutropenia. Journal of Immunology (Baltimore, Md.: 1950). 2015;195(4):1341–1349.
- Cheers C, Haigh AM, Kelso A, et al. Production of colony-stimulating factors (CSFs) during infection: separate determinations of macrophage-, granulocyte-, granulocyte-macrophage-, and multi-CSFs. Infect Immun. 1988;56(1):247–251.
- Kawakami M, Tsutsumi H, Kumakawa T, et al. Levels of serum granulocyte colony-stimulating factor in patients with infections. Blood. 1990;76(10):1962–1964.
- Hartung T. Effect of filgrastim treatment on inflammatory cytokines and lymphocyte functions. Clin Pharmacol Ther. 1999;66(4):415–424.
- Glogauer M. L Goldman, AI Schafer, editor. 172 - Disorders of phagocyte function, in goldman's cecil Medicine (Twenty fourth edition). Philadelphia: W.B. Saunders; 2012. p. 1111–1118.
- Lord BI, Molineux G, Pojda Z, et al. Myeloid cell kinetics in mice treated with recombinant interleukin-3, granulocyte colony-stimulating factor (CSF), or granulocyte-macrophage CSF in vivo. Blood. 1991;77(10):2154–2159.
- Basu S, Hodgson G, Katz M, et al. Evaluation of role of G-CSF in the production, survival, and release of neutrophils from bone marrow into circulation. Blood. 2002;100(3):854–861.
- Roskos LK, Lum P, Lockbaum P, et al. Pharmacokinetic/pharmacodynamic modeling of pegfilgrastim in healthy subjects. J Clin Pharmacol. 2006;46(7):747–757.
- Chen W, Boras B, Sung T, et al. A physiological model of granulopoiesis to predict clinical drug induced neutropenia from in vitro bone marrow studies: with application to a cell cycle inhibitor. J Pharmacokinet Pharmacodyn. 2020;47(2):163–182.
- Chen T-L, Chiang Y-W, Lin G-L, et al. Different effects of granulocyte colony-stimulating factor and erythropoietin on erythropoiesis. Stem Cell Res Ther. 2018;9(1):1–9.
- Modi J, Menzie-Suderam J, Xu H, et al. Mode of action of granulocyte-colony stimulating factor (G-CSF) as a novel therapy for stroke in a mouse model. J Biomed Sci. 2020;27(1):1–19.
- Nicola NA, Metcalf D. Binding of the differentiation-inducer, granulocyte-colony-stimulating factor, to responsive but not unresponsive leukemic cell lines. Proc Natl Acad Sci U S A. 1984;81(12):3765–3769.
- Boneberg E-M, Hareng L, Gantner F, et al. Human monocytes express functional receptors for granulocyte colony-stimulating factor that mediate suppression of monokines and interferon-gamma. Blood. 2000;95(1):270–276.
- Morikawa K, Morikawa S, Nakamura M, et al. Characterization of granulocyte colony-stimulating factor receptor expressed on human lymphocytes. Br J Haematol. 2002;118(1):296–304.
- Harada M, Qin Y, Takano H, et al. G-CSF prevents cardiac remodeling after myocardial infarction by activating the Jak-stat pathway in cardiomyocytes. Nat Med. 2005;11(3):305–311.
- Bussolino F, Wang JM, Defilippi P, et al. Granulocyte- and granulocyte– macrophage-colony stimulating factors induce human endothelial cells to migrate and proliferate. Nature. 1989;337(6206):471–473.
- Schneider A, Krüger C, Steigleder T, et al. The hematopoietic factor G-CSF is a neuronal ligand that counteracts programmed cell death and drives neurogenesis. J Clin Invest. 2005;115(8):2083–2098.
- McCracken S, Layton JE, Shorter SC, et al. Expression of granulocyte-colony stimulating factor and its receptor is regulated during the development of the human placenta. J Endocrinol. 1996;149(2):249–258.
- Roberts AW. G-CSF: a key regulator of neutrophil production, but that's not all!. Growth Factors. 2005;23(1):33–41.
- Manz MG, Miyamoto T, Akashi K, et al. Prospective isolation of human clonogenic common myeloid progenitors. Proc Natl Acad Sci USA. 2002;99(18):11872–7.
- Fukunaga R, Ishizaka-Ikeda E, Nagata S. Purification and characterization of the receptor for murine granulocyte colony-stimulating factor. J Biol Chem. 1990;265(23):14008–14015.
- Layton JE, Hall NE. The interaction of G-CSF with its receptor. Front Biosci. 2006;11:3181–3189.
- Touw IP, van de Geijn GJ. Granulocyte colony-stimulating factor and its receptor in normal myeloid cell development, leukemia and related blood cell disorders. Front Biosci. 2007;12:800–815.
- Wright CR, Ward AC, Russell AP. Granulocyte colony-stimulating factor and Its potential application for skeletal muscle repair and regeneration. Mediators Inflamm. 2017;7517350:1–9.
- Baker SJ, Rane SG, Reddy EP. Hematopoietic cytokine receptor signaling. Oncogene. 2007;26(47):6724–6737.
- Fukunaga R, Seto Y, Mizushima S, et al. Three different mRNAs encoding human granulocyte colony-stimulating factor receptor. Proc Natl Acad Sci USA. 1990;87(22):8702–8706.
- Liongue C, Wright C, Russell AP, et al. Granulocyte colony-stimulating factor receptor: stimulating granulopoiesis and much more. Int J Biochem Cell Biol. 2009;41(12):2372–2375.
- Ward AC, Hermans MHA, Smith L, et al. Tyrosine-dependent and -independent mechanisms of STAT3 activation by the human granulocyte colony-stimulating factor (G-CSF) receptor are differentially utilized depending on G-CSF concentration. Blood. 1999;93(1):113–124.
- Larsen A, Davis T, Curtis BM, et al. Expression cloning of a human granulocyte colony-stimulating factor receptor: a structural mosaic of hematopoietin receptor, immunoglobulin, and fibronectin domains. J Exp Med. 1990;172(6):1559–1570.
- Mehta HM. Alternatively spliced, truncated GCSF receptor promotes leukemogenic properties and sensitivity to JAK inhibition. Leukemia. 2014;28(5):1041–1051.
- Tamada T. Homodimeric cross-over structure of the human granulocyte colony-stimulating factor (GCSF) receptor signaling complex. Proc Natl Acad Sci U S A. 2006;103(9):3135–3140.
- Liu, L., et al. The role of granulocyte colony-stimulating factor in breast cancer development: A review. Molecular medicine reports. 2020;21(5):2019–2029.
- Ward AC, Smith L, de Koning JP, et al. Multiple signals mediate proliferation, differentiation, and survival from the granulocyte colony-stimulating factor receptor in myeloid 32D cells. J Biol Chem. 1999;274(21):14956–14962.
- Hermans MHA, Antonissen C, Ward AC, et al. Sustained receptor activation and hyperproliferation in response to granulocyte colony-stimulating factor (G-CSF) in mice with a severe congenital neutropenia/acute myeloid leukemia-derived mutation in the G-CSF receptor gene. J Exp Med. 1999;189(4):683–692.
- Dwivedi P, Greis KD. Granulocyte colony-stimulating factor receptor signaling in severe congenital neutropenia, chronic neutrophilic leukemia, and related malignancies. Exp Hematol. 2017;46:9–20.
- Geissler K, Gunzer M, Ostermann H. How safe Is the administration of long-acting granulocyte colony-stimulating factor in cancer patients? Oncol Res Treat. 2018;41(5):316–326.
- Demetri GD, Griffin JD. Granulocyte colony-stimulating factor and its receptor. Blood. 1991;78(11):2791–2808.
- Corey SJ, Burkhardt AL, Bolen JB, et al. Granulocyte colony-stimulating factor receptor signaling involves the formation of a three-component complex with Lyn and Syk protein-tyrosine kinases. Proc Natl Acad Sci U S A. 1994;91(11):4683–4687.
- Dwivedi P, Greis KD. Granulocyte colony-stimulating factor receptor signaling in severe congenital neutropenia, chronic neutrophilic leukemia, and related malignancies. Exp Hematol. 2017;46:9–20.
- Nguyen-Jackson H, Panopoulos AD, Zhang H, et al. STAT3 controls the neutrophil migratory response to CXCR2 ligands by direct activation of G-CSF-induced CXCR2 expression and via modulation of CXCR2 signal transduction. Blood. 2010;115(16):3354–3363.
- Nguyen-Jackson HT, Li HS, Zhang H, et al. G-CSF-activated STAT3 enhances production of the chemokine MIP-2 in bone marrow neutrophils. J Leukoc Biol. 2012;92(6):1215–1225.
- Palande K. Scratching the surface: signaling and routing dynamics of the CSF3 receptor. Front Biosci. 2013;18:91–105.
- Zeidler C, Schwinzer B, Welte K. Congenital neutropenias. Rev Clin Exp Hematol. 2003;7(1):72–83.
- Hermans MHA, van de Geijn G-J, Antonissen C, et al. Signaling mechanisms coupled to tyrosines in the granulocyte colony-stimulating factor receptor orchestrate G-CSF-induced expansion of myeloid progenitor cells. Blood. 2003;101(7):2584–2590.
- Touw IP, van de Geijn G-JM. Granulocyte colony-stimulating factor and its receptor in normal myeloid cell development, leukemia and related blood cell disorders. Front Biosci. 2007;12:800–815.
- Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol. 2008;8(7):533–544.
- Souza LR. G-CSF activation of AKT is not sufficient to prolong neutrophil survival. J Leukoc Biol. 2013;93(6):883–893.
- Bajrami B, Zhu H, Kwak H-J, et al. G-CSF maintains controlled neutrophil mobilization during acute inflammation by negatively regulating CXCR2 signaling. J Exp Med. 2016;213(10):1999–2018.
- Martin C, Burdon PCE, Bridger G, et al. Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity. 2003;19(4):583–593.
- Burdon PC, Martin C, Rankin SM. The CXC chemokine MIP-2 stimulates neutrophil mobilization from the rat bone marrow in a CD49d-dependent manner. Blood. 2005;105(6):2543–2548.
- Eash KJ, Greenbaum AM, Gopalan PK, et al. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J Clin Invest. 2010;120(7):2423–2431.
- Pelus LM, Fukuda S. Peripheral blood stem cell mobilization: the CXCR2 ligand GRObeta rapidly mobilizes hematopoietic stem cells with enhanced engraftment properties. Exp Hematol. 2006;34(8):1010–1020.
- Panopoulos AD, Bartos D, Zhang L, et al. Control of myeloid-specific integrin alpha mbeta 2 (CD11b/CD18) expression by cytokines is regulated by Stat3-dependent activation of PU.1. J Biol Chem. 2002;277(21):19001–7.
- Bajrami B, Zhu H, Kwak H-J, et al. G-CSF maintains controlled neutrophil mobilization during acute inflammation by negatively regulating CXCR2 signaling. J Exp Med. 2016;213(10):1999–2018.
- Pelus LM, Horowitz D, Cooper SC, et al. Peripheral blood stem cell mobilization. Crit Rev Oncol Hematol. 2002;43(3):257–275.
- Devi S, Wang Y, Chew WK, et al. Neutrophil mobilization via plerixafor-mediated CXCR4 inhibition arises from lung demargination and blockade of neutrophil homing to the bone marrow. J Exp Med. 2013;210(11):2321–2336.