
ABSTRACT
Objective: The 8p11 myeloproliferative syndrome [EMS] is a rare myeloproliferative disorder which usually develops rapidly with chromosomal translocation of the fibroblast growth factor receptor 1 gene. The gene has 15 fusion partners, including the breakpoint cluster region (BCR) gene on chromosome 22. Of all the tests available, chromosome karyotype determination is the most important for the diagnosis of EMS. Here, we describe one case of a patient characterized by marked increase of white blood cells and thrombocytopenia and diagnosed as EMS with t(8;22)(p11;q11) by chromosome karyotype.
Methods: 28-year-old man was referred to our hospital. He had a onemonth history of intermittent coughing and a small amount of expectoration after catching a cold. As an outpatient, his complete blood count showed: WBC was 130.04 × 109/L with 80.20% granulocytes.Hematologic investigations, bone marrow analysis and genomic DNA sequencing studies were performed.
Results: Despite additional chromosomal abnormalities,the patient progressed rapidly with a B blast cell clone in one month. After diagnosis inthree months, the patient underwent the haplo-identical BMT of his brother, followed up for three years, and had a high rate of survival.
Conclusions: Our report provides a definite conceptual framework for a better understanding of the characteristics of The 8p11 myeloproliferative syndrome [EMS].
1. Introduction
The 8p11 myeloproliferative syndrome [EMS] is a rare myeloproliferative disorder that usually progresses rapidly with chromosomal translocation of the fibroblast growth factor receptor1 (FGFR1) gene. EMS may present as myeloid hyperplasia, eosinophilia, and a high incidence of T-lymphoblastic lymphoma. The FGFR1 gene is located at chromosome 8p11 and may fuse with different partner genes [Citation1–5].
In 2001, Fioretos reported a new fusion gene involving BCR and FGFR1 genes in 8p11 myelodysplastic syndrome EMS [Citation2]. Since then, 14 additional cases of this rare fusion have been reported, all presenting as atypical chronic myelogenous leukemia (CML) rather than typical EMS. All were more likely to develop B lymphoblastic lymphoma, mixed phenotype B/myeloid acute leukemia, or even third-age T/B/myeloid hybrid acute leukemia [Citation6]. Patients with 8P11 myelodysplastic syndrome with BCR–FGFR1 gene rearrangement had a poor prognosis despite intensive chemotherapy. Allogeneic hematopoietic stem cell transplantation (HSCT) is considered the only treatment option.
Here, we detail the case t(8;22)(p11;q11) and BCR/FGFR1 rearrangement in a rapid progressing B blast cell clone without additional chromosomal abnormalities. Three months after diagnosis, the patient underwent the haplo-identical BMT of his brother, followed up for three years, and had a high rate of survival.
2. Case report
2.1. Clinical data
In May 2018, a 28-year-old man was referred to Bao’an District People Hospital in Shenzhen. He had a one-month history of intermittent coughing and a small amount of expectoration after catching a cold. As an outpatient, his complete blood count showed: WBC was 130.04 × 109/L with 80.20% granulocytes. Hemoglobin was 119 g/L and platelet count was 30 × 109/L. Since his blood was abnormal, the patient was admitted to the hospital for further inspection. The patient was formerly physically healthy. He denied night sweats, sleepiness, anorexia or the desire to lose weight. He did not smoke or drink. However, his uncle previously had leukemia. Physical examination was negative for hepatosplenomegaly. Cervical, axillary and inguinal lymph nodes were not palpable.
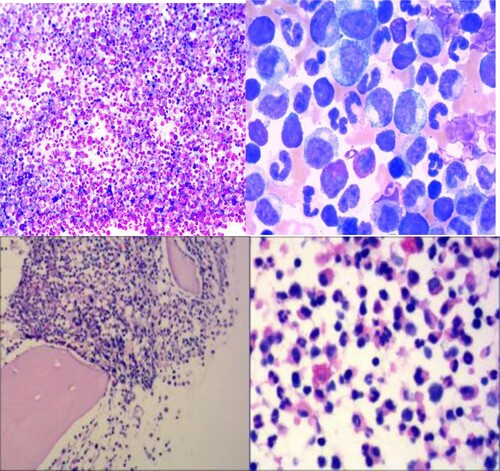
During hospitalization, the patient underwent bone marrow-related examinations. Bone marrow aspirates showed hyperproliferative granulocytes (73.5%), no excess protocells (0%) and no dysplasia. Basophil (0%) and eosinophil (3%) did not expand. Twenty-six megakaryocytes were found and all were granular without thromocytogenic. Granulocytes at various stages were seen in peripheral blood smears, including promyelocyte, myelocyte and metamylocyte. Peripheral blood smear showed basophils (0%), eosinophil (1.5%) promyelocytes (1%), myelocytes (5.5%) and metamyelocytes (16%). The morphological manifestations are similar to change terminology to CML of chronic phase (). Bone marrow examination revealed mild hypercellular of marrow with granulocytic hyperplasia. Eosinophils and basophils were not evident. The first flow cytology results were as follows: a flow cytometry analysis of bone marrow cells revealed an 86.5% granulocyte system account without dysplastic features, and 3.5% blast cell expressed as CD34, HLA-DR and Myeloperoxidase (MPO). Lymphocytes account for 2.5%, and they were polyclonal. The second flow cytology results in one month later: Flow cytometric analysis showed that the protocells (2%) were positive for HLA-DR, CD10(bri), CD38(dim), CD19 and particallyCD34. MPO was negative. These observations suggested that the blast cells were phenotypically immature B lymphocytes. A bone marrow biopsy showed a large proliferation of granular cells. Abnormal Localization of Immature Precursor was present, and eosinophils were easy to see. It showed the megakaryocytes presented as hyperplasia, and small and monolobate megakaryocytes were present without bone marrow fibrosis. The morphological manifestations were similar to MPN.
Figure 1. (A) Bone marrow aspirate: numerous granulocytes without dysplasia. Wright stain (100×). (B) Wright stain (1000×). (C) Bone marrow biopsy: hypercellular particle with granulocytic predominance. Hematoxylin and eosin stain (100×) and (D) hematoxylin and eosin stain (400×).
The 20 metaphase bone marrow cells showed 46,XY, t(8; 22) (p11; q11), no other abnormal karyotytes were found. ((A)) An interphase fluorescence in situ hybridization (FISH) study using the FGFR1 probes revealed division signals in 96% of the cells. The BCR/ABL double fusion probe identified the translocation partner of BCR in metaphase FISH analysis and showed the splitting signal of the BCR probe (three green BCR gene signals and two red ABL gene signals) in 92% of the cells. ((B)) The mutation of CSF3R gene (exon 1-17) was detected by second-generation sequencing. Intron mutation was detected at site c.485 + 71A > G (heterozygous sum). According to the current database, the mutation was a single nucleotide polymorphic site and was not pathogenic. Based on these results, the patients were diagnosed with BCR–FGFR1 gene rearrangement of 8p11 myeloproliferative syndrome.
Figure 2. (A) Chromosome analysis by G banding showing inserted karyotype of 46,XY,t(8;22) (p11;q11) in 20 out of 20 metaphases with no other abnormal karyotytes in this patient. (B) The fusion signal is observed in the patient by FISH. nuc ish (FGFR1×2)(5′FGFR1 sep 3′FGFR1×1)[384/400].
![Figure 2. (A) Chromosome analysis by G banding showing inserted karyotype of 46,XY,t(8;22) (p11;q11) in 20 out of 20 metaphases with no other abnormal karyotytes in this patient. (B) The fusion signal is observed in the patient by FISH. nuc ish (FGFR1×2)(5′FGFR1 sep 3′FGFR1×1)[384/400].](/cms/asset/aac48eb6-da96-42fd-b2a9-59bcc4b25e89/yhem_a_1971889_f0002_oc.jpg)
2.2. Treatment and prognosis
During hospitalization, the patient received initial treatment with hydroxyurea. The patient had intermittent epistaxis when platelets significantly decreased and thus was treated with EItrombopag 50 mg QD to promote platelet growth. Two months later, the patient developed hip pain without difficulty walking. The patient underwent an MRI examination. The MRI showed widespread bone signal abnormalities in lumbar vertebrae, bilateral upper femur and pelvic bones, patchy enhancement of right iliac bone, acetabulum and left femoral head, and a small amount of effusion in bilateral hip joints. This suggested bone involvement. On 25 August, the patient underwent haploid HSCT with HLA matching 5/10. While considering the patient’s economic condition, we used FISH for regular follow-ups (once every six months). After three years of follow-up, the patient’s blood was normal, and there was no obvious GVHD.
3. Discussion
Myeloproliferative syndrome (EMS) is a kind of entity proposed by Macdonald et al. in 1995 for a case of 8p11 translocation [Citation7]. These patients exhibited clinicopathologic similarities, including a high incidence of myeloproliferative syndrome with eosinophilia, lymphadenopathy and T-cell lymphoblastic lymphoma progressing to acute myeloid leukemia. EMS is considered to be a rare invasive tumor associated with translocation or insertion of the fibroblast growth factor receptor 1 (FGFR1) gene on chromosome 8P11-12. This resulted in the production of a new gene rearrangement and in the expression of a chimeric protein with conformational activation of its FGFR1 tyrosine kinase domain [Citation8]. EMS was defined in the 2008 World Health Organization classification [Citation9] as ‘myeloid and lymphoid tumors with FGFR1 abnormalities’. BCR is one of the 15 fusion partners of FGFR1. The 15 rearrangement karyotypes are as follows: Myosin XVIIIA (MYO18A; 17q23), tripartite motif containing 24 (TIF1; 7q34), FGFR1 oncogene partner 2 (FGFR1OP2; 12p11), human endogenous retrovirus group K member (HERV–K; 19q13), BCR (22q11), centriolin (CEP110; 9q33), FGFR1 oncogene partner (FOP/FGFR10P; 6q27), zinc finger protein 198 (ZNF198; 13q11–12), cleavage and polyadenylation specific factor 6 (CPSF6; 12q15), tripartite motif containing 24 (TRIM24; 7q34), nuceloporin 98 (NUP98; 11p15), cut like homeobox 1 (CUX1; 7q22), translocated promoter region, nuclear basket protein(TPR; 1q25), RAN binding protein 2 (RANBP2; 2q12) and LPR binding FLII interacting protein 1 (LRRFIPI; 2q37) [Citation8]. In patients with FGFR1 rearrangement, t(8; 22)(p11; q11) and BCR/FGFR1 fusion genes have clinical characteristics more similar to chronic myeloid leukemia (CML) than classical EMS [Citation5], or there is less acute B lymphoblastic leukemia [Citation10]. It has been reported in the literature the t(8;22)(p11.2;q11.2)/BCR–FGFR1 may arise from a myeloid/B progenitor cell. It is important to recognize that neoplasms carrying the t(8;22)/BCR–FGFR1, although rare, can commonly with B lymphoblastic leukemia at the initial diagnosis [Citation11].
The clinical characteristics of EMS were not characteristic. It could be manifested as hypermetabolic symptoms, fever, fatigue, weakness, weight loss and night sweating. But not all of the patients had these symptoms. Most of the patients showed increased peripheral blood leukocytes, mainly neutrophils and some eosinophils [Citation12]. The morphology of bone marrow in most patients was not significantly different from that of typical CML. In this case, and also in the previously reported several cases, no other additional chromosomal abnormalities were observed except for t(8;22). These results underscore the importance of cytogenetics and molecular analysis in patients with atypical CML. Subsequently, the detection of genes involved in the tyrosine kinase pathway may become an increasingly important feature of diagnosis [Citation13].
Professor Meng reported one case of t(8;22)(p11;q11) and BCR/FGFR1 rearrangement without extra chromosomal abnormalities, which presented with an inert disease process, followed up for two years without progress [Citation14]. Unlike Professor Meng’s case, the clinical course of the same chromosomal abnormality in our report is greatly different. The B blast cell clone in the bone marrow in one month was detected in our patient, and it progressed rapidly. It was speculated that different molecular mutations might be involved in disease progression.
The course of EMS also had chronic and acute stages. In the chronic phase, myeloid cells proliferate, but they could still differentiate and mature, and the blast cells in bone marrow were less than 5%. Without effective treatment, the patient quickly progressed to an acute stage. At present, the duration of the chronic phase could not be accurately predicted. Most patients would progress within one to two years after diagnosis. The median conversation period was about six to nine months, which was greatly different from CML. The latter had a chronic period of two to seven years. The disease can develop into acute leukemia, usually myeloid or mixed cells, with a small number of T or B cells [Citation8]. Even if EMS patients were given intensive chemotherapy, which reduced the enlarged liver and spleen, chromosome abnormalities still persisted. It was difficult to achieve complete remission. The median survival time was 12 months. Long-term remission is maintained in a small number of patients receiving HSCT. At present, allogeneic HSCT was the only promising treatment for long-term remission of EMS patients.
Tyrosine kinase inhibitors (TKIs) have been shown to be successful in the treatment of CML. The research has proved that the third-generation TKI ponatinib has clear activity against FGFR1 fusion proteins in EMS. Thus it has the potential to treat this rare subset of MPN patients for whom current therapy is inadequate [Citation15], and different TKIs have been suggested as alternatives to the treatment of 8p11 myelodysplastic syndrome [Citation16]. Recently, it has been reported that a patient with BCR/FGFR1 translocation obtained a clinical response to ponatinib treatment [Citation17]. Although currently the only therapeutic option is considered to be allogeneic HSCT, TKIs may provide a therapeutic option for patients who do not meet the criteria for allogeneic HSCT. Or, it can bridge the gap between diagnosis and allogeneic HSCT [Citation16]. In addition, TKI maintenance therapy after allogeneic HSCT may also have a beneficial effect on clinical outcomes of allogeneic HSCT from unrelated HLA-mismatched donors by reducing the risk of GvHD [Citation18]. Because the disease is rare, there are few reports of TKI inhibitor in 8p11 application. But in this patient, we did not use TKI before or after allogeneic HSCT.
In the treatment of EMS, a large number of studies have reported that allogeneic HSCT can benefit patients [Citation18,Citation19]. Through matched bone marrow transplantation or umbilical cord blood transplantation, some EMS patients received remission and long-term survival. We reported one patient who underwent transplantation three months after diagnosis. The donor was his brother, 5/10 of whom was matched. After following up with the patient for three years, he has survived well.
Previous literature reported 11 cases of bone marrow transplantation with different degrees of remission. Currently, the continuous improvement of the transplantation system and the continuous application of half-matched bone marrow transplantation achieve the same therapeutic effect of full-matched donors. Donors were no longer a problem for most EMS patients [Citation20,Citation21]. Patients with high-risk myeloid malignancies may benefit from a prophylactic haplo-DLI, which should ideally be used in the setting of a clinical trial [Citation22].
This case was a 28-year-old male with no symptoms of hypermetabolism, such as fever, night sweats, or weight loss. This patient only had coughing, which lasted for two months. The blood routine was characterized by high white blood cells and low platelets. The cytological and histopathological examination of bone marrow shows marked proliferation of granulocytes. CML was initially suspected. No abnormal myeloid blast cells and lymphocytes were found in the first report of immunotyping of hematological tumors, which suggested marked proliferation of granulocytes. Two percent abnormal B lymphocytes were found in the second report of immunotyping of hematological tumors after reexamining for one month, which suggested clonal evolution of lymphocytes. After a series of molecular biology, CML, MPN, and CMML were excluded. At last, cytogenetics and FISH were further confirmed as 8p11 syndrome. The patient had no superficial lymph nodes, no hepatosplenomegaly, and no lymph node biopsy. However, the patient developed hip pain without difficulty walking, and underwent an MRI examination. It was found that widespread bone signal abnormalities in lumbar vertebrae, bilateral upper femur and pelvic bones, patchy enhancement of right iliac bone, acetabulum and left femoral head, and a small amount of effusion was found in bilateral hip joints. This suggested bone involvement. During hospitalization, hydroxyurea was given to reduce cells and leukocytes decreased, but platelets remained at about 30 × 109/L. They were maintained by platelet transfusion. Chromosome abnormalities were always present and could not be alleviated genetically. The patient underwent transplantation three months after diagnosis. The donor was his brother, with 5/10 HLA locus match. After following up for three years, the patient had a good rate of survival.
In conclusion, EMS is a rare malignant hematological disease with different clinical manifestations. Diagnosis needed to exclude other myeloproliferative diseases. It was easy to misdiagnose. Cytogenetics and molecular biology techniques were necessary. After diagnosis, clinical stages (chronic or acute stages) were required. Combined with the comprehensive monitoring of MICM, it can detect the progress of the disease in time. At present, the prognosis of EMS is still poor; chemotherapy alone was ineffective and cytogenetic remission could not be achieved. Although many laboratory and clinical data suggest that new targeted drugs have great application prospects in the treatment of EMS, they have not yet been formally applied in the clinic. Currently, HSCT is still an effective method to treat EMS, and early HSCT can enable patients to obtain long-term survival.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Demiroglu A, Steer EJ , Heath C, et al. The t(8;22) in chronic myeloid leukemia fuses BCR to FGFR1: transforming activity and specific inhibition of FGFR1 fusion proteins. Blood 2001;98(13):3778–3783.
- Fioretos T, Panagopoulos I , Lassen C, et al. Fusion of the BCR and the fibroblast growth factor receptor-1 (FGFR1) genes as a result of t(8;22)(p11;q11) in a myeloproliferative disorder: the first fusion gene involving BCR but not ABL. Genes, Chromosomes Cancer 2001;32(4):302–310.
- Pini M, Gottardi E, Scaravaglio P, et al. A fourth case of BCR-FGFR1 positive CML-like disease with t(8;22) translocation showing an extensive deletion on the derivative chromosome 8p. Hematol J 2002;3(6):315–316.
- Murati A, Arnoulet C, Lafage-Pochitaloff M, et al. Dual lympho-myeloproliferative disorder in a patient with t(8;22) with BCR-FGFR1 gene fusion. Int J Oncol 2005;26(6):1485–1492.
- Richebourg S, Theisen O, Plantier I, et al. Chronic myeloproliferative disorder with t(8;22)(p11;q11) can mime clonal cytogenetic evolution of authentic chronic myelogeneous leukemia. Genes, Chromosomes Cancer 2008;47(10):915–918.
- Khodadoust MS, Luo B, Medeiros BC, et al. Clinical activity of ponatinib in a patient with FGFR1-rearranged mixed-phenotype acute leukemia. Leukemia 2016;30(4):947–950.
- Macdonald D, Aguiar RC, Mason P, et al. A new myeloproliferative disorder associated with chromosomal translocations involving 8p11: a review. Leukemia 1995;9(10):1628–1630.
- Jackson CC, Medeiros LJ, Miranda RN. 8p11 myeloproliferative syndrome: a review. Hum Pathol 2010;41(4):461–476.
- Sabattini E, Bacci F, Sagramoso C, et al. WHO classification of tumours of haematopoietic and lymphoid tissues in 2008: an overview. Pathologica 2010;102(3):83–87.
- Baldazzi C, Iacobucci I, Luatti S, et al. B-cell acute lymphoblastic leukemia as evolution of a 8p11 myeloproliferative syndrome with t(8;22)(p11;q11) and BCR-FGFR1 fusion gene. Leuk Res 2010;34(10):e282–e285.
- Montenegro-Garreaud X, Miranda RN, Reynolds A, et al. Myeloproliferative neoplasms with t(8;22)(p11.2;q11.2)/BCR-FGFR1: a meta-analysis of 20 cases shows cytogenetic progression with B-lymphoid blast phase. Hum Pathol 2017;65:147–156.
- Antic DA, Vukovic VM, Milosevic Feenstra JD, et al. 8p11 myeloproliferative syndrome: diagnostic challenges and pitfalls. J Buon 2016;21(3):745–749.
- Qin YW, Yang YN, Bai P, et al. Chronic myelogenous leukemia-like hematological malignancy with t(8;22) in a 26-year-old pregnant woman: a case report. Oncol Lett 2016;11(6):4131–4133.
- Liu JJ, Meng L. 8p11 myeloproliferative syndrome with t(8;22)(p11;q11): a case report. Exp Ther Med 2018;16(2):1449–1453.
- Chase A, Bryant C, Score J, et al. Ponatinib as targeted therapy for FGFR1 fusions associated with the 8p11 myeloproliferative syndrome. Haematologica 2013;98(1):103–106.
- Landberg N, Dreimane A, Rissle M, et al. Primary cells in BCR/FGFR1-positive 8p11 myeloproliferative syndrome are sensitive to dovitinib, ponatinib, and dasatinib. Eur J Haematol 2017;99(5):442–448.
- Khodadoust MS, Luo B, Medeiros BC, et al. Clinical activity of ponatinib in a patient with FGFR1-rearranged mixed-phenotype acute leukemia. Leukemia 2016;30(4):947–950.
- Konishi Y, Kondo T, Nakao K, et al. Allogeneic hematopoietic stem cell transplantation for 8p11 myeloproliferative syndrome with BCR-FGFR1 gene rearrangement: a case report and literature review. Bone Marrow Transplant 2018;54:326–329.
- Morishige S, Oku E, Takata Y, et al. A case of 8p11 myeloproliferative syndrome with BCR-FGFR1 gene fusion presenting with trilineage acute leukemia/lymphoma, successfully treated by cord blood transplantation. Acta Haematol 2013;129(2):83–89.
- Lv M, Chang Y, Huang X. Everyone has a donor: contribution of the Chinese experience to global practice of haploidentical hematopoietic stem cell transplantation. Front Med 2018;54:45–56.
- Xu L, Chen H, Chen J, et al. The consensus on indications, conditioning regimen, and donor selection of allogeneic hematopoietic cell transplantation for hematological diseases in China-recommendations from the Chinese society of hematology. J Hematol Oncol 2018;11(1):33.
- Dholaria B, Savani BN, Labopin M, et al. Clinical applications of donor lymphocyte infusion from an HLAhaploidentical donor: consensus recommendations from the acute leukemia working party of the EBMT. Haematologica 2020;105(1):47–58.