
ABSTRACT
Objective
IDH1/2 mutations, intervening in epigenetic procedures, are frequently encountered in acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS). Knowledge of the genetics, immunophenotypes, and mutational kinetics of IDH1/2-mutated AML can contribute to the understanding of AML clonal architecture and inform therapeutics and monitoring.
Methods
We retrospectively analyzed 50 IDH1/2-mutated AML/MDS-EB cases of our institution, to identify recurrent co-mutations, immunophenotypes, patterns of co-variance of IDH1/2 allele burdens with those of recurrent co-mutations, frequency of persistent IDH1/2 mutation as clonal hematopoiesis of indeterminate potential (CHIP) in remission and response to hypomethylating agents.
Results
Most frequently co-mutated genes were DNMT3A, SRSF2 and NPM1. Most cases with co-existent IDH1/2 and NPM1 mutations (11/13) showed an ‘APL-like’ immunophenotype (CD34-HLADR-). Allele burdens of mutated IDH1/2 were identical to mutated SRSF2 allele burdens at diagnosis and remission, but not always to mutated NPM1 allele burden in remission. We show persistence of significant mutIDH1/2 allele burden in approximately one-fourth of patients with deep remissions. IDH1/2 mutations were significantly more frequent among responders to first-line HMA-based regimens than among non-responders, in patients treated for myeloid neoplasms with excess blasts.
Conclusions
IDH1/2 mutations are most frequently accompanied by DNMT3A, SRSF2 and NPM1 mutations. NPM1-IDH1/2 mutated AML has a mature phenotype possibly amenable to differentiation therapies. IDH1/2 and SRSF2 mutations probably arise at the same developmental stage of the disease, as their allele burdens covariate. IDH1/2 mutation represents CHIP in a substantial proportion of cases and is therefore no reliable residual disease marker. The preferential presence of IDH1/2 mutations among HMA-responders could inform therapeutic decisions if confirmed in larger series.
Introduction
IDH (isocitrate dehydrogenase) enzymes convert, via a two-step process, isocitrate to α-ketoglutarate (α-KG), within and outside of, the Krebs cycle [Citation1]. α-KG produced from isocitrate can serve as a substrate (OH-donor) for dioxygenase-type enzymes. Dioxygenase-type enzymes include, among others, histone demethylases and DNA-5-methylcytosine hydroxylases like TET2. Therefore, as hydroxylation of the 5-methylcytosines of DNA is the first step to DNA demethylation [Citation2,Citation3], the activity of IDH1/2 enzymes, ultimately serves, among other purposes, to promote DNA and histone demethylation by providing to dioxygenases the substrate α-KG, and thus to activate certain gene expression signatures [Citation4]. Mutations of IDH1/2 are frequent in gliomas and cartilaginous tumors [Citation5,Citation6] and occur recurrently in AML and MDS [Citation7]. IDH1 mutations typically alter the R132 residue of the active site, while mutations in IDH2 similarly alter an active-site arginine at position 140 or 172 [Citation4,Citation7]. Disturbance of the active site leads to production of D-2-hydroxyglutarate (D-2-HG), instead of α-KG, a product with ability to inhibit dioxygenase-type enzymes, instead of serving as their substrate [Citation8–10]. Therefore, both histone and DNA demethylation are ultimately inhibited by the non-physiological product D-2-HG and a hypermethylated genome/epigenome is expected, and has been shown, in IDH1/2-mutated malignancies [Citation11–13].
In AML, the IDH1/2 enzymes are mutated with a cumulative frequency of about 20% (7% for IDH1 and 13% for IDH2, with mutations changing usually the R140 and less frequently the R172 residue of IDH2) and these mutations are potentially early lesions in the oncogenic event cascade [Citation7,Citation14]. Existing data show that in vitro transfection of murine bone marrow cells with mutant IDH1/IDH2 constructs impairs hematopoietic differentiation and increases stem/progenitor cell marker expression [Citation12], while IDH1-R132Q ‘knock-in’ mice show anemia and myeloproliferative features [Citation15]. Transgenic mice with conditional expression of IDH2-R140Q also present impaired erythroid maturation and, again, a proliferative trait of extramedullary hematopoiesis, while expression of the mutant transgene contributes to the manifestation of leukemia in the presence of additional lesions such as exogenous HoxA9 overexpression [Citation16]. Lethal myelodysplasia with proliferative features arises in animal models expressing simultaneously mutated IDH2 and mutated spliceosome factor SRSF2, something not seen in the relevant work with either mutation alone [Citation17].
Questions of clinical and scientific importance for IDH1/2-mutated AML consist in the identification of recurrent co-existing mutations or chromosomal abnormalities, the definition of mutations happening at the same oncogenic stage with IDH1/2 as opposed to ‘upstream’ or ‘downstream’ mutations, the phenotypes of IDH1/2-mutated AML, the phenotypes defined by specific co-existing mutations, the kinetics of IDH1/2 mutational burden in remission and relapse and the presence or not of a relapse risk related to persistence of clonal hematopoiesis with IDH1/2 mutations in remission. Of interest is also whether the presence of IDH1/2 mutations favors a response to hypomethylating agents (HMAs), given their mechanism of action, as well as these mutations’ influence on the outcome of distinct genetic categories of AML, such as AML with NPM1 mutation.
We address some of these questions via retrospective evaluation of our institutional cohort of patients with AML and MDS-EB (MDS with excess blasts) with IDH1/2 mutations.
Methods
Patients
We identified 50 patients with AML/ MDS-EB diagnosed in our institution from 01.01.2014 to 31.03.2022 (43 AML, 6 MDS-EB and 1 blastic plasmacytoid dendritic cell neoplasm) carrying IDH1 or IDH2 mutations or both (). Sixteen patients had an IDH1 mutation, 33 an IDH2 mutation and 1 patient had both. We did not include cases of AML falling into any of the WHO 2017 categories of ‘AML with recurrent genetic abnormalities’, except AML with mutation of NPM1, which has been described to be frequent in cases with IDH1/2 mutation [Citation18]. This is because recurrent chromosomal abnormalities (such as the translocations of CBF-leukemias or KMT2A translocations) are central to the pathogenesis of the corresponding AML cases and would be the predominant factors affecting the phenotype, concurrent lesions and outcome. Interestingly, among our patients with IDH1/2 mutations, there was only one case falling into these categories (one with a KMT2A-MLLT3 fusion), which was excluded from the cohort.
Table 1. Mutation sites, demographic and hematologic characteristics, diagnosis, first-line treatment and response rates of our cohort of AML/MDS-EB patients with IDH1/2 mutations diagnosed since 2014 (WH0 2017 nomenclature).
We moreover collected data of all patients with myeloid malignancies with excess blasts treated first-line with an hypomethylating agent (HMA) in our institution from 01.01.2018 to 31.03.2022, to compare the proportion of IDH1/2-mutated cases between responders and non-responders (the IDH1/2-mutated patients of this series were part of our 50-patient cohort of IDH1/2-mutated patients). Twenty-seven patients were identified and characterized as responders if hematologic CR/CRi was achieved, and as non-responders if CR/CRi was not achieved after at least 3 cycles of HMA or HMA + venetoclax treatment ().
Table 2. Patients with myeloid malignancies with excess blasts, treated 1st-line with hypomethylating agents. Responders: achieving hematologic CR or CRi (notably all of them achieved CR). Non-responders: not achieving CR/CRi after at least 3 cycles of treatment.
Flow cytometry
Flow cytometry at diagnosis was performed using combinations of fluorochrome-conjugated antibodies against CD38, CD34, HLA-DR, CD117, CD13, CD33, CD123, CD4, CD56, CD64, CD14, CD300e, CD61, CD15, CD65, cMPO, CD19, CD10, cCD3, CD3, CD7, cTdT, cCD79a. Cases with blast population negative for both CD34 and HLA-DR (expression less than 1 log) were defined as "double-negative" myeloid phenotype ("DN"), also known as "acute-promyelocytic leukemia-like" phenotype ("APL-like"). All other cases were named as "non-DN" phenotype. All cases of the cohort fulfilled WHO criteria for myeloid or monocytic lineage of the blasts. Sensitivity of our flow cytometry follow-up of residual disease is usually at 0.01%.
Oncogenetics analyses
All cases were analyzed at diagnosis by: conventional karyotyping or array-CGH; RT-MLPA for fusion transcripts with previously published methods [Citation19]; PCR with FRET probes and melting curve analysis for NPM1 mutations; PCR and electrophoretic migration on capillary sequencer for the presence of FLT3-ITD; quantitative WT1 gene expression (with overexpression defined as ratio WT1 copies/104 ABL1 copies >250 in bone marrow); and NGS panel containing at least the genes ASXL1, DNMT3A, CEBPA, IDH1, IDH2, KIT, NPM1, RUNX1 and TP53 (this being the limited NGS panel used in 2014–2015). For cases diagnosed since 2016, the NGS panel became more comprehensive to include in total 35 genes from 2018 onwards (ASXL1, BCOR, BRAF, CALR, CBL, CEBPA, CSF3R, DNMT3A, ETV6, EZH2, FLT3, GATA2, HRAS, IDH1, IDH2, JAK2, KIT, KRAS, MPL, NPM1, NRAS, PHF6, PRPF8, PTPN11, RUNX1, SETBP1, SF3B1, SH2B3, SRSF2, STAG2, TET2, TP53, U2AF1, WT1, ZRSR2) (Oncomine TM Myeloid Research Assay, Fisher Scientific, MA, USA; Seqpilot Module SeqNext versions 4.3-5.0, JSI Medical Systems GmbH, Ettenheim, Germany; Alamut, IARC). Mutated genes’ variant allele frequencies (VAF) are measured in this cohort by NGS, with an error margin of 1-2%. Quantitative NPM1-mutation follow-up was performed by classical quantitative RT–PCR, with results given as ratio of expression to the gene ABL1.
Remission definitions/Statistics
Comparison of the incidence (odds ratio) of "DN" phenotype among groups with the presence or absence of each of the most prevalent co-mutations (NPM1, SRSF2, any codon DNMT3A) was performed via the Fisher’s exact test. Testing for a significant difference of the mutated IDH1/2 allele burden (VAF, with value 0–100% or 0–1.0) from the mutated NPM1 and SRSF2 allele burdens at diagnosis and in remission was performed with paired Wilcoxon tests (testing the statistical significance of each one of the differences IDH1/2-NPM1 VAF and IDH1/2-SRSF2 VAF at diagnosis and in remission). Estimation of confidence intervals for the proportion of patients with IDH1/2 mutation persistence (>2% VAF IDH1/2 on NGS) in "deep CR/CRi" was performed with the binomial proportion formula. We define "deep CR/CRi" as hematologic CR/CRi with maximum 0.1% myeloblasts on flow cytometry; higher blast percentage (always respecting hematologic CR) is allowed for cases with blasts of nonspecific phenotype and a sensitive molecular marker proving remission (WT1 expression normal and reduced more than 2 log for cases with initial WT1 overexpression; <0.1% mutated NPM1 and reduced at least 3-log for NPM1-mutated AML). A comparison of the frequency of IDH1/2 mutations between responders (hematologic CR/CRi) and non-responders to HMA-based first-line treatment was performed with the Fisher’s exact test.
Results
Results for the presence of coexisting gene mutations at diagnosis are summarized in . DNMT3A mutations were the most frequent, present in 23/50 patients, of whom 8 IDH1-mutated, 14 IDH2-mutated and 1 double-mutated IDH1/2 patient. Twenty different DNMT3A mutations overall were detected, with five patients carrying more than one DNMT3A mutation and with six patients carrying the active site-inactivating hotspot R882 mutation. Second most-frequent mutation was the SRSF2 (a spliceosome factor [Citation20,Citation21]) P95 mutation, overall present in 16/50 patients, 13 of whom were IDH2-mutated and 3 IDH1-mutated. Third most frequent mutation was the NPM1 W288Cfs*12 mutation, present in 13/50 patients, of whom 6 with an IDH1 mutation, 6 with an IDH2 mutation and 1 with both mutations. Regarding karyotypic or array-detected genetic abnormalities, most common ones were 7q abnormalities, found in 8/50 patients, and trisomy 8, present in 5/50 patients. There were 5 cases with complex karyotype; the most frequent karyotype was normal, present in 27/47 patients (karyotype not available in three patients of the series).
Figure 1. Each column corresponds to one patient. Red cages signal that the horizontally corresponding mutation is present in the respective patient; the symbols * or # within a red cage denote that 2 or 3 different mutations of the corresponding gene are present. Gray cages signify that the corresponding gene was not analyzed in the respective patient. DNMT3A mutations (including the hotspot R882 codon mutations, which are shown with an H, and numerous others), SRSF2 codon P95 mutations and NPM1 W288Cfs*12 mutations are the most frequent in the cohort.

Analysis of the immunophenotype of our IDH1/2-mutated AML/MDS-EB cases showed a highly significant correlation between the simultaneous presence of IDH1/2 and NPM1 mutations and a "double-negative" myeloid phenotype (A), as first named by Mason and colleagues [Citation22], meaning a myeloid phenotype lacking both CD34 and HLA-DR (‘acute promyelocytic leukemia-like’) and highly expressing MPO and CD33 (example of this phenotype in suppl. ). Indeed, out of the 13 NPM1-mutated cases of our series, 11 presented a DN phenotype (11/13), compared to 0/37 for the NPM1-wild type cases (odds ratio confidence interval: 22.06 – ∞). There was no correlation between the presence/absence of the other two most frequent mutations (SRSF2 P95 mutations and DNMT3A mutations) with the phenotype being DN or not (B & C).
Table 3. The presence of the NPM1 mutation in IDH1/2 mutated patients is highly correlated with a double-negative ("DN" or "APL-like") immunophenotype. No significant correlations of co-existing SRSF2 or DNMT3A mutations with phenotype.
Mutational load (VAF by NGS) of IDH1/2 at diagnosis was not always equivalent to the mutational load of NPM1 in our cohort cases. A paired Wilcoxon test could not, however, reject equivalence of IDH1/2 and NPM1 VAFs at diagnosis (p-value = 0.18). In remission, though, the same test rejected equivalence of IDH1/2 and NPM1 VAFs (p = 0.022), as in numerous cases the mutational load of IDH1/2 was much higher than that of NPM1 ((A)); notably, the NPM1 mutational load on NGS was zero for all our patient cases, for which data were available, in remission. On the contrary, we observed the VAF of IDH1/2 mutation to be equal in all cases to the VAF of SRSF2 mutation, both at diagnosis and in remission (the difference IDH1/2 minus SRSF2 VAF in both states being non-significant in the paired Wilcoxon tests, (B)). We did not investigate the significance of the differences of the DNMT3A mutations’ VAFs with IDH1/2 VAFs, due to the considerable heterogeneity of DNMT3A mutations.
Figure 2. Boxplots of the variant allele frequencies (VAFs) expressed as values 0.0–1.0. A. IDH1/2 and NPM1 VAFs at diagnosis and remission, designated as d and r, respectively: there is a significant difference between IDH1/2 and NPM1 VAFs in remission in paired Wilcoxon testing, originating from the fact that certain patients keep high IDH1/2 VAF in remission. Tendency for difference at diagnosis without significance proven. B. IDH1/2 and SRSF2 VAFs at diagnosis and remission show no differences, with certain patients keeping high VAFs of both mutations in remission. C. Of the 31 patients for whom a ‘deep CR’ was documented (definition in ‘Methods’), 7/31 had persistent IDH1/2 VAF > 2% during deep remission (noise threshold in NGS assay). Duration of follow-up is given in months. Timepoints of measurements are shown with circles. Patients 3.2, 11.2 and 25.2 were NPM1-mutated and showed undetectable NPM1-mutated transcript during follow-up, on qPCR, with sensitivity 0.01%.
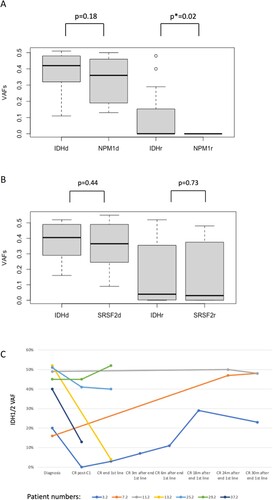
Concerning the persistence, or disappearance, of IDH1/2 mutations, in hematologic CR, we naturally evaluated for this purpose only CRs obtained before/without allografting: of the 45 patients who were both able to receive treatment and followed-up for responses, 41/45 achieved hematologic CR/CRi with first of subsequent line of treatment before eventual allografting (37 of them with one or two cycles of induction chemotherapy and 4 of them with HMA-based regimen) (). At least 31 of these 41 patients had a confirmed "deep CR/CRi", as defined in the Methods:Statistics section. Seven of these 31 patients had persistent IDH1/2 VAF > 2% in "deep CR/CRi" (2% being the noise threshold in our NGS assay); consequently, we discover approximately 23% of patients in deep remission to be carrying persistent IDH1/2 mutations, with range of VAFs 4%-52%. The evolution of the VAFs of mutated IDH1/2 for these seven patients in deep remission is shown at different timepoints during follow-up in (C). Three of the seven patients (3/7) with persistent IDH1/2 mutation in "deep CR/CRi" happened to be NPM1-mutated and were, noteworthy to say, in complete molecular remission during follow-up, evidenced by undetectable normalized NPM1-mutated transcript with sensitivity 0.01%. None of the seven patients relapsed or lost their MRD-negative remission in the period of observation available for each, with longest periods of observation (before allograft or with no subsequent allograft) being 30 months after the end of consolidation, for three of the patients (the rest of the patients had shorter follow-ups of the IDH1/2 VAF, as they were either transplanted or followed-up in other centers).
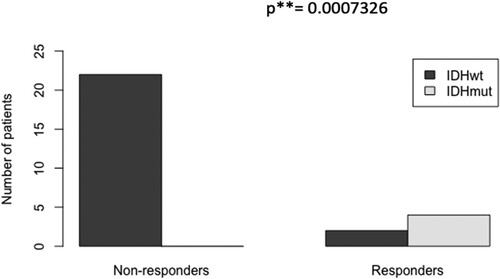
As IDH1/2 mutations are thought to contribute to the pathogenesis of myeloid disease by promoting aberrant DNA or histone methylation or both, we investigated if, among all the azacitidine- and decitabine-first-line-treated patients (with myeloid neoplasms with excess blasts) since 2018 in our institution, IDH1/2 mutations were more frequent in responders than in non-responders. Characteristics of the patients are shown in . As shown in , IDH1/2 mutations were significantly more frequent in the group of responders (present in 4/6) in comparison to the group of non-responders (0/21) (odds ratio ∞, p < 0.001 in Fisher’s exact test).
Figure 3. The proportion of IDH1/2-mutated cases among responders to HMA-based treatment (1st-line) was significantly higher than among non-responders (4/6 vs 0/21, p < 0.001, characteristics of patients shown in ).
Discussion
In our institutional series of 50 IDH1/2-mutated patients with AML and MDS-EB, excluding AML with recurrent genetic abnormalities other than AML with NPM1 mutation, DNMT3A was the most frequently co-mutated gene, at various nucleotide positions. A co-occurrence of IDH1/2 and DNMT3A mutations has been noted before [Citation23–25] and an elegant work with mouse models showed synergy of the two mutations for leukemic transformation via H3K9 hypermethylation and suggested that leukemia with loss of both DNMT3A and IDH2 may be amenable to therapeutic targeting by histone deacetylase inhibitors [Citation26]. Spliceosome factor SRSF2 was the second most frequently mutated gene in our cohort, always at position P95. The unequivocal preferential association of IDH1/2 (most frequently IDH2) and SRSF2 codon 95-specific mutations has not been adequately highlighted in the literature, although frequent co-occurrence was mentioned in two studies [Citation24,Citation27]; an interesting work with murine models transduced with a double mutant IDH2/SRSF2 vector showed indeed that spliceosome dysfunction is a critical mediator in the process of IDH2 mutation-driven leukemogenesis [Citation17,Citation28]. Third most common gene mutation in our cohort was the NPM1 mutation. Frequent co-occurrence with IDH1/2 mutations has been described [Citation25], but mechanisms of potential synergy are unknown; a relevant work has shown that hypoxia-driven pathways are activated in the presence of both mutations [Citation29]. A mutual exclusivity of TET2-mutations with IDH1/2 (mutations acting probably by the same mechanism of decrease of DNA dioxygenase activity) has been reported [Citation30,Citation31]; our series includes, however, one case with coexistent TET2 mutation. Regarding chromosomal abnormalities in our patient population, most prevalent was normal karyotype, present in 57% of cases, and most common anomaly was 7q deletion, present in 16% of cases. A higher prevalence of intermediate-risk cytogenetics among IDH1/2-mutated leukemias has been observed in the literature [Citation7].
Almost all cases (11/13) of IDH1/2-mutated NPM1-mutated AML in our patient population had a CD34- HLADR- phenotype, named "double-negative" or "APL-like", whereas none of the IDH1/2-mutated cases without NPM1 mutation manifested this phenotype. This statistically significant observation is in accordance with a previous report, which, inversely, showed that 96–99% of NPM1-mutated leukemias with "double-negative" phenotype harbor IDH1/2 or TET2 mutations [Citation22]. As mentioned earlier, TET2 mutations act by disrupting the same mechanism of DNA demethylation [Citation30], which could explain the similarity of phenotypes between IDH1/2- and TET2-mutated cases with NPM1 mutation in the relevant work. As a reminder, approximately a third of NPM1-mutated AML have a monocytic phenotype, whereas the rest show a myeloid phenotype, of which about half show an "APL-like" myeloid phenotype, which means no expression of the stem cell marker CD34 and of HLA-DR and strong expression of MPO [Citation22,Citation32]. The mechanism by which the co-presence of NPM1 and IDH1/2 or TET2 mutations favors this acute promyelocytic leukemia-like phenotype is unclear. This phenotype may imply that these leukemias, similarly to APL, stem from a ‘mature’ leukemic cell and could possibly be driven to differentiation via novel therapeutic approaches additional to chemotherapy (such as ATRA or IDH1/IDH2 inhibitors) or could benefit from alleviated chemotherapy regimens associated to these agents. Indeed, Mason and colleagues showed clearly superior outcomes for NPM1-mutated AML with double-negative phenotype [Citation32]. Moreover, one preclinical work has shown that AML cell lines with IDH1 R132 are prone to differentiation when exposed to pharmacological doses of ATRA [Citation33]. Interesting clinical data show a differentiation syndrome (DS) in 12-40% of the patients after administration of the IDH2 inhibitor enasidenib and in up to 40% of patients for the IDH1-inhibitor ivosidenib in R/R AML, a complication more frequently seen in cases with high blast percentage [Citation34,Citation35]. Whether there is a correlation of the development of DS with immunophenotype (‘APL-like’) or genetics (co-presence of NPM1 mutation in the respective cases) has not yet been investigated.
Regarding the correlation of the allele burden of IDH1/2 with those of the commonly coexisting mutations, accordance of allele frequencies is seen in our series between IDH1/2 mutation and SRSF2 mutation, both at diagnosis and in remission, which implies that IDH1/2 and SRSF2 mutations are invariably present in the same clone. On the contrary, there is frequent discordance of allelic burden of IDH1/2 with NPM1 in remission (and sometimes at diagnosis; statistical analysis cannot reject equality of the two allele frequencies at diagnosis, but p-value is marginal) which implies that NPM1 mutation is often subclonal to IDH1/2 mutations (the allele burden of mutIDH1/2 in the NPM1-mutated cases concerned being higher than that of mutNPM1, (A)). This finding implies that IDH1/2 mutations often represent CHIP (clonal hematopoiesis of indeterminate potential), whereas an NPM1 mutation may be acquired later in the course of the disease, within a preexisting CHIP clone.
A significant proportion of patients (7 of the 31 cases who achieved confirmedly ‘deep complete remission’ according to our definition, which gives a confidence interval of 10–41% given the size of our population) had considerable persistent mutated IDH1/2 allele burdens in remission, with three of these cases being even in complete molecular remission documented by the absence of NPM1 mutation by sensitive methods. This finding shows, again, that IDH1/2 mutation kinetics do not follow disease evolution in all cases, with the mutation sometimes representing CHIP. The proportion of IDH1/2 mutation representing CHIP in our cohort seems therefore to be approximately 1 in 5 at least. In conclusion, IDH1/2 should not be considered as an MRD marker for newly diagnosed cases and its potential use for this purpose must be evaluated case by case considering the patient's entire cytogenetic and mutational profile and their kinetics [Citation36]. Similar results have been obtained in one cohort published during the course of our work, whereas a French group reported that in 7% of patients with AML with IDH1/2 mutations, these persisted at high levels in complete remission, consistent with the presence of an IDH1/2 mutation in a pre-leukemic hematopoietic stem cell [Citation37,Citation38].
It is uncertain whether or not the persistence of a pre-leukemic clone with IDH1/2 mutation in deep remission confers an increased relapse risk, but it is reasonable to hypothesize that, in deep remission of the initial disease, acquisition of novel mutations from the persisting CHIP clone is needed for the re-emergence of myeloid disease and thus the risk of ‘relapse’ is merely equivalent to the risk of evolution into a new myeloid neoplasia of the corresponding CHIP. Three of our patients with persistence of IDH1/2 mutations in CR have been followed for periods longer than 2.5 years with no evidence of relapse despite high IDH1/2 allele burdens ranging 23% to 48%. Indeed, another research group described nine cases of AML with persistent IDH1/2 mutations in CR, none of whom relapsed or developed novel myeloid disease in periods of observation of 7–52 months [Citation39]. On the contrary, Debarri and colleagues showed, that, in a small cohort of NPM1-mutated IDH1/2-mutated patients, out of four patients with discordant NPM1 and IDH1/2 quantifications in remission (NPM1 mutation ≤0.1% by qPCR and IDH1/2 VAF ≥ 5% on NGS), three of them relapsed in intervals 158–504 days post-diagnosis and one evolved into an NPM1-negative MDS; it is noteworthy, however, that all three relapsing cases had detectable NPM1 transcripts in remission, even if only 0.01–0.02% [Citation40]. Ok and colleagues showed a significantly higher relapse risk (59% vs 24%) for patients with persistent versus non-persistent IDH1/2 mutation in CR/CRi in a cohort of 80 AML patients treated by intensive chemotherapy, but the depth of cytogenetic/molecular remissions of these patients is not mentioned [Citation41].
Complete response rate to first-line treatment was high in our series (41/45), with only four cases of first-line failure; comparison of the response rate in the different co-mutation or treatment subsets was not attempted due to lack of power. Similarly, our patient population lacks critical size and duration of follow-ups to analyze relapse-free or overall survival in the different subsets or even in its whole, given the relatively increased censoring for loss to follow-up. Interestingly, a multicentric study has recently shown that patients with IDH1 R132C mutation have a lower CR rate and a trend for reduced overall survival compared to other mutant IDH1 variants and to wild-type IDH1/2 [Citation42]. On the contrary, patients with IDH2 R172K in the ELN intermediate/adverse-risk group in the same study (too few patients with IDH2-R172K mutation belonged to the favorable risk group) had significantly better relapse-free and overall survival than wild-type IDH1/2 patients in the same group, whereas IDH2-R140Q mutations did not have prognostic significance for this unfavorable risk population. Overall, prognostic impact of IDH1/2 mutations in AML is far from being unequivocally established, with several controversial outcomes, especially in smaller studies [Citation18] and possibly depends on the type of IDH mutation and largely on the concomitant genetic profile. One study clearly showed that the presence of NPM1 mutation is the primary prognostic factor for overall survival in IDH1-mutated and IDH2 R140-mutated AML treated by intensive chemotherapy [Citation43].
We show that among patients with myeloid neoplasms treated first-line in our institution by HMA-based regimens, IDH1/2 mutations are significantly more frequent in responders (CR/CRi), present in 67% of them, in comparison to 0% of non-responders. The good response of IDH1/2-mutated myeloid neoplasms to hypomethylating agents may be related to the postulated disease-provoking mechanism of IDH/2 mutations (disruption of DNA-demethylation). The presence of IDH1/2 mutations could thus inform treatment decisions in chemotherapy-unfit patients. We did not assess the durability of responses to HMAs of IDH1/2-mutated cases versus other cases. Our results are in accordance with a previous work, which, reversely, looked if IDH1/2-mutated AML has a higher response rate to HMAs than IDH1/2 wild-type AML and indeed found a significant odds ratio of response of 14.2 in favor of the IDH1/2-mutated cases [Citation44]. During the analysis of our data, the group of the VIALE-A study [Citation45] reported superior overall survival for IDH1/2-mutated AML treated first-line with azacitidine and venetoclax, compared to IDH1/2 wild-type AML treated with the same regimen, but did not show significant differences in remission rates [Citation46]. Recent trials of the combination of HMAs with IDH1/2 inhibitors show a 100% CR rate for first-line treatment of newly diagnosed IDH2-mutated AML cases unfit for chemotherapy by enasidenib and azacitidine, whereas the combination of ivosidenib and azacitidine is promising for IDH1-mutated AML, with one-year event-free survival approaching 40% [Citation47,Citation48].
In conclusion, our retrospective study shows that IDH1/2 mutations, epigenetic modifiers acting through disruption of histone modifications and DNA hypermethylation, are preferentially associated with SRSF2 codon P95 mutations, NPM1 mutation and various DNMT3A mutations. The mechanisms of synergy of IDH1/2 mutations with these genetic alterations remain to be identified, to provide insight into AML pathogenesis and disease targeting. Leukemic blasts doubly mutated for NPM1, and IDH1/2, show an "APL-like" phenotype (HLADR- and CD34-), which implies a mature leukemic population possibly amenable to differentiation therapies. IDH1/2 mutation allele burden always coincides and co-variates with the SRSF2 mutation burden, implying that they arise at the same developmental stage of the disease; this is not true for the NPM1 mutation. IDH1/2 mutations persist as clonal hematopoiesis in profound complete remission in at least one fifth of our cases and their persistence does not herald increased risk for disease relapse in our cohort, as patients followed up longer than two years did not relapse despite high IDH1/2 allele burdens. The CR rate of IDH1/2-mutated myeloid neoplasms was high in our patient series. IDH1/2 mutations were much more frequent in responders versus in non-responders to HMA-based treatment in our institution.
Author contributions
V.P. conceived the study, collected and analyzed data and wrote the manuscript. J.P.S and F.S. supervised genetics analyses and interpretation. V.B. supervised immunophenotyping and interpretation. J.P. analyzed data. S.B. reviewed the manuscript. O.S. synthetically analyzed data and reviewed the manuscript.
Supplemental Material
Download MS Word (455.1 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Corpas FJ, Barroso JB, Sandalio LM, et al. Peroxisomal NADP-Dependent isocitrate dehydrogenase. Characterization and activity regulation during natural senescence. Plant Physiol. 1999;121(3):921–928.
- Ploumakis A, Coleman ML. OH, the places you’ll go! Hydroxylation, gene expression, and cancer. Mol Cell. 2015;58(5):729–741.
- Dong C, Zhang H, Xu C, et al. Structure and function of dioxygenases in histone demethylation and DNA/RNA demethylation. IUCrJ. 2014;1(Pt 6):540–549.
- Garrett-Bakelman FE, Melnick AM. Mutant IDH: a targetable driver of leukemic phenotypes linking metabolism, epigenetics and transcriptional regulation. Epigenomics. 2016;8(7):945–957.
- Yang H, Ye D, Guan KL, et al. IDH1 and IDH2 mutations in tumorigenesis: mechanistic insights and clinical perspectives. Clin Cancer Res. 2012;18(20):5562–5571.
- Huang J, Yu J, Tu L, et al. Isocitrate dehydrogenase mutations in Glioma: from basic discovery to therapeutics development. Front Oncol. 2019;9:506.
- DiNardo CD, Ravandi F, Agresta S, et al. Characteristics, clinical outcome, and prognostic significance of IDH mutations in AML. Am J Hematol. 2015;90(8):732–736.
- Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2010;465(7300):966.
- Losman JA, Kaelin WG. What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013;27(8):836–852.
- Xu W, Yang H, Liu Y, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19(1):17–30.
- Lu C, Ward PS, Kapoor GS, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483(7390):474–478.
- Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553–567.
- Wilson ER, Helton NM, Heath SE, et al. Focal disruption of DNA methylation dynamics at enhancers in IDH-mutant AML cells. Leukemia. 2022;36(4):935–945.
- Cazzola M. IDH1 and IDH2 mutations in myeloid neoplasms–novel paradigms and clinical implications. Haematologica. 2010;95(10):1623–1627.
- Sasaki M, Knobbe CB, Munger JC, et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature. 2012;488(7413):656–659.
- Kats LM, Reschke M, Taulli R, et al. Proto-oncogenic role of mutant IDH2 in leukemia initiation and maintenance. Cell Stem Cell. 2014;14(3):329–341.
- Yoshimi A, Lin KT, Wiseman DH, et al. Coordinated alterations in RNA splicing and epigenetic regulation drive leukaemogenesis. Nature. 2019;574(7777):273–277.
- Medeiros BC, Fathi AT, DiNardo CD, et al. Isocitrate dehydrogenase mutations in myeloid malignancies. Leukemia. 2017;31(2):272–281.
- Ruminy P, Marchand V, Buchbinder N, et al. Multiplexed targeted sequencing of recurrent fusion genes in acute leukaemia. Leukemia. 2016;30(3):757–760.
- Thol F, Kade S, Schlarmann C, et al. Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood. 2012;119(15):3578–3584.
- Arbab Jafari P, Ayatollahi H, Sadeghi R, et al. Prognostic significance of SRSF2 mutations in myelodysplastic syndromes and chronic myelomonocytic leukemia: a meta-analysis. Hematology. 2018;23(10):778–784.
- Mason EF, Kuo FC, Hasserjian RP, et al. A distinct immunophenotype identifies a subset of NPM1-mutated AML with TET2 or IDH1/2 mutations and improved outcome. Am J Hematol. 2018;93(4):504–510.
- Hou HA, Kuo YY, Liu CY, et al. DNMT3A mutations in acute myeloid leukemia: stability during disease evolution and clinical implications. Blood. 2012;119(2):559–568.
- Lin CC, Hou HA, Chou WC, et al. IDH mutations are closely associated with mutations of DNMT3A, ASXL1 and SRSF2 in patients with myelodysplastic syndromes and are stable during disease evolution. Am J Hematol. 2014;89(2):137–144.
- Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209–2221.
- Zhang X, Wang X, Wang XQD, et al. Dnmt3a loss and Idh2 neomorphic mutations mutually potentiate malignant hematopoiesis. Blood. 2020;135(11):845–856.
- Pollyea DA, Tallman MS, de Botton S, et al. Enasidenib, an inhibitor of mutant IDH2 proteins, induces durable remissions in older patients with newly diagnosed acute myeloid leukemia. Leukemia. 2019;33(11):2575–2584.
- Yen K, Travins J, Wang F, et al. AG-221, a first-in-class therapy targeting acute myeloid leukemia harboring oncogenic IDH2 mutations. Cancer Discov. 2017;7(5):478–493.
- Ogawara Y, Katsumoto T, Aikawa Y, et al. Kitabayashi I: IDH2 and NPM1 mutations cooperate to activate Hoxa9/Meis1 and Hypoxia pathways in acute myeloid leukemia. Cancer Res. 2015;75(10):2005–2016.
- Gurnari C, Pagliuca S, Guan Y, et al. TET2 mutations as a part of DNA dioxygenase deficiency in myelodysplastic syndromes. Blood Adv. 2022;6(1):100–107.
- Chan SM, Majeti R. Role of DNMT3A, TET2, and IDH1/2 mutations in pre-leukemic stem cells in acute myeloid leukemia. Int J Hematol. 2013;98(6):648–657.
- Mason EF, Hasserjian RP, Aggarwal N, et al. Blast phenotype and comutations in acute myeloid leukemia with mutated NPM1 influence disease biology and outcome. Blood Adv. 2019;3(21):3322–3332.
- Boutzen H, Saland E, Larrue C, et al. Isocitrate dehydrogenase 1 mutations prime the all-trans retinoic acid myeloid differentiation pathway in acute myeloid leukemia. J Exp Med. 2016;213(4):483–497.
- Fathi AT, DiNardo CD, Kline I, et al. Investigators A-C-S: differentiation syndrome associated with Enasidenib, a selective inhibitor of mutant isocitrate dehydrogenase 2: analysis of a phase 1/2 study. JAMA Oncol. 2018;4(8):1106–1110.
- Norsworthy KJ, Mulkey F, Scott EC, et al. Differentiation syndrome with ivosidenib and enasidenib treatment in patients with relapsed or refractory IDH-mutated AML: a U.S. Food and Drug Administration systematic analysis. Clin Cancer Res. 2020;26(16):4280–4288.
- Schuurhuis GJ, Heuser M, Freeman S, et al. Minimal/measurable residual disease in AML: a consensus document from the European LeukemiaNet MRD Working Party. Blood. 2018;131(12):1275–1291.
- Kovy P, Orfi Z, Bors A, et al. Nucleophosmin1 and isocitrate dehydrogenase 1 and 2 as measurable residual disease markers in acute myeloid leukemia. PLoS One. 2021;16(6):e0253386.
- Ferret Y, Boissel N, Helevaut N, et al. Clinical relevance of IDH1/2 mutant allele burden during follow-up in acute myeloid leukemia. A study by the French ALFA group. Haematologica. 2018;103(5):822–829.
- Wiseman DH, Williams EL, Wilks DP, et al. Frequent reconstitution of IDH2(R140Q) mutant clonal multilineage hematopoiesis following chemotherapy for acute myeloid leukemia. Leukemia. 2016;30(9):1946–1950.
- Debarri H, Lebon D, Roumier C, et al. IDH1/2 but not DNMT3A mutations are suitable targets for minimal residual disease monitoring in acute myeloid leukemia patients: a study by the Acute Leukemia French Association. Oncotarget. 2015;6(39):42345–42353.
- Ok CY, Loghavi S, Sui D, et al. Persistent IDH1/2 mutations in remission can predict relapse in patients with acute myeloid leukemia. Haematologica. 2019;104:305–311.
- Middeke JM, Metzeler KH, Rollig C, et al. Differential impact of IDH1/2 mutational subclasses on outcome in adult AML: results from a large multicenter study. Blood Adv. 2021;6(5):1394–1405.
- Duchmann M, Micol JB, Duployez N, et al. Prognostic significance of concurrent gene mutations in intensively treated patients with IDH-mutated AML: an ALFA study. Blood. 2021;137(20):2827–2837.
- Emadi A, Faramand R, Carter-Cooper B, et al. Presence of isocitrate dehydrogenase mutations may predict clinical response to hypomethylating agents in patients with acute myeloid leukemia. Am J Hematol. 2015;90(5):E77–E79.
- DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and Venetoclax in previously untreated acute Myeloid Leukemia. N Engl J Med. 2020;383(7):617–629.
- Pollyea DA, DiNardo CD, Arellano ML, et al. Impact of Venetoclax and Azacitidine in treatment-naive patients with acute myeloid leukemia and IDH1/2 mutations. Clin Cancer Res. 2022;28(13):2753–2761.
- Venugopal S, Takahashi K, Daver N, et al. Efficacy and safety of enasidenib and azacitidine combination in patients with IDH2 mutated acute myeloid leukemia and not eligible for intensive chemotherapy. Blood Cancer J. 2022;12(1):10.
- Montesinos P, Recher C, Vives S, et al. Ivosidenib and Azacitidine in IDH1-mutated acute myeloid leukemia. N Engl J Med. 2022;386(16):1519–1531.