
ABSTRACT
Objective: The clinical and genetic characteristics of a child with inherited bone marrow failure syndrome as prominent clinical manifestations and special facial features were analyzed, and the etiology and mechanism were explored in, combination with clinical practice. Methods: Blood samples and clinical information were collected separately from the proband and their biological parents. The pathogenic variant was verified using next-generation sequencing technology screening, and the candidate variable sites were confirmed by using Sanger sequencing among all members of the family. Results: A heterozygous nonsense mutation in exon 17 of KAT6A (NM_006766), c.4177G > T (p.E1393*) predicted to cause truncation within the acidic domain of the protein was identified. Pedigree analysis did not reveal any variation in this locus between the proband's father and mother. No report of this pathogenic variant was found in a literature search of domestic and foreign databases, indicating that it is a newly discovered mutation. According to the guidelines of the American College of Medical Genetics, the variation was preliminarily determined to be a pathogenic. The newly discovered heterozygous mutation in KAT6A may be the cause of the disease in this child. Additionally, inherited bone marrow failure syndrome is a prominent manifestation. Conclusion: This study not only provides us with an in-depth understanding of this rare syndrome but also deepens our understanding of the function of KAT6A.
1. Introduction
Lysine acetyltransferase 6A (KAT6A), also known as MOZ or MYST3 among other names, belongs to the MYST family. The MYST family is one of four protein families (MYST family, p300/ CBP family, GCN5/PCAF family and Rtt109 family) distinguished by amino acid sequence homology with lysine acetyltransferases (KATs). The main catalytic substrates are nuclear matrix proteins, and histones such as H3, H4, and H2A. It performs epigenetic regulation by altering the histone acetylation levels [Citation1]. Although this protein family is evolutionarily conserved, KAT6A is unique to vertebrates [Citation2]. Human KAT6A is located on chromosome 8, 8q11, and contains 17 exons, of which exon 1 is the smallest and exon 17 is the largest. The abnormal function of KAT6A resulting from genetic mutations may affect the protein levels and activities of the genes it regulates, and multiple systems or organs can be affected. Currently, this condition is clinically defined KAT6A syndrome.
KAT6A syndrome is very rare, with 334 known cases as of October 2021 according to statistics from the KAT6A Foundation. However, owing to insufficient knowledge and diverse clinical phenotypes, the estimated number of misdiagnosed or undiagnosed patients is difficult to precisely determine.
KAT6A syndrome is named after the gene harbouring the disease-causing mutation. The KAT6A gene(chr8-41791561) belongs to a group of genes that can cause monogenic neurodevelopmental disorders [Citation3]. Being a relatively recent discovery, very few cases of KAT6A syndrome have been reported both nationally and internationally. The most common manifestations of KAT6A syndrome include intellectual disability or developmental delay with relatively distinct facial features, often involving the eyes, ears, heart, and digestive system, along with behavioral abnormalities and sleep disturbances [Citation4]. Hematological involvement has rarely been reported. With respect to clinical phenotypes involving the hematological system, several cases of isolated neutropenia have been previously reported in the literature [Citation5], in addition to one case of persistent thrombocytopenia and several cases of severe infection with suspected immunodeficiency [Citation6]. Patients with KAT6A syndrome are easily misdiagnosed or underdiagnosed due to a lack of unique and unifying features that allow for easy recognition by physicians. Given the rarity and common misdiagnosis of this syndrome, the relationship between genotype and phenotype has not yet been fully elucidated. As the syndrome is considered a Mendelian disorder of the epigenetic machinery [Citation7], influenced from both background genetic variation and the environment may overlap, result in complexity in the clinical phenotypes. Correlation studies of newly discovered mutations and clinical phenotypes may be an the optimal research strategy at this stage. We hope to use this as an opportunity to further explore the underlying mechanisms of this syndrome and to guide clinical treatment.
2. Materials and methods
2.1 Clinical presentation
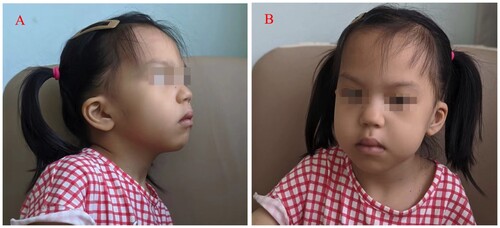
A 4-year-old female child was admitted to the hospital after a diagnosis of ‘pancytopenia for more than 4 years.’ She was born through a Caesarean section at full term as her parents’ second child. Her parents’ phenotypes were normal and they denied any family history of hereditary disease. The child was found to have pancytopenia a few days after birth, and her intellectual and psychomotor development was behind peers of the same age a few months after birth. A bone marrow biopsy was performed at a foreign hospital in an attempt to diagnose her ‘congenital aplastic anemia?’, and initial genetic testing did not reveal any clinically significant mutations. Hematopoietic stem cell transplantation was refused, therefore, intermittent blood transfusion was required. There was an episode of a transient increase in liver enzymes, and the patient had two episodes of pneumonia and two episodes of infectious diarrhea accompanied by thickening of the intestinal wall, both of which were cured using antibiotic treatment. At the current age of 4 years, the child can only verbalize ‘mama.’ She is able to understand sentences used during daily communication and express herself using simple body movements. Her gross and fine motor development is age appropriate. While her cognitive level is slightly behind, the child’s personality is relatively lively. Distinct facial features are present in the skull and jaws, including a broad nasal tip, bitemporal narrowing, epicanthic folds, low-set posteriorly rotated ears, a short and flattened philtrum, a tented upper lip, downturned corners of the mouth, and mild micrognathia, but no microcephaly. () The child’s head circumferences during infancy and when she was a toddler were also largely appropriate for children of the same age, and there was no premature closure of the anterior fontanelle. The child had slight strabismus, but no significant abnormalities in her visual field were found after the ophthalmologic examination. Congenital structural heart anomalies are another relatively common phenotype. In the present case, a ventricular septal defect was found immediately after birth. There were no feeding difficulties or gastroesophageal reflux, but there were two episodes of transient intestinal wall thickening.
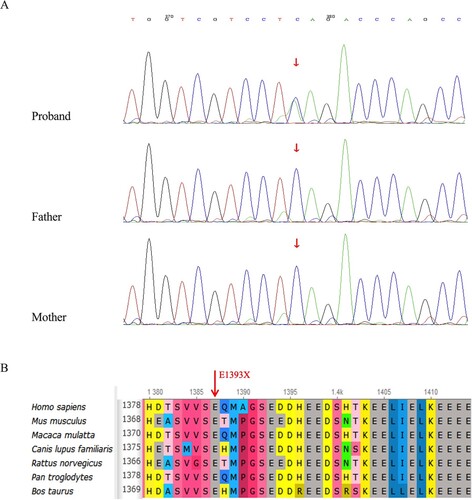
Figure 1. Results of gene sequencing and conservation analysis. (A) De novo variant of KAT6A (variant c.4177G>T). (B) conservation analysis of mutation site.
2.2 Methodology
2.2.1 Sequencing and bioinformatics
Genomic DNA was isolated from the peripheral blood samples from the patient and her parents. Trio-whole exome sequencing was performed by MyGenostics (Beijing, China) using a nova6000 sequencing platform. After sequencing, low-quality variants were filtered using a quality score threshold of ≥20. Filtered fragments were compared to the reference human genome (hg19) using Burrows–Wheeler Aligner software. Single nucleotide polymorphisms and insertions or deletions were analyzed using the Genome Analysis Toolkit. After the above steps, the data were converted to the VCF format, and variants were further annotated using ANNOVAR and associated with multiple databases such as 1000 Genomes, ESP6500, dbSNP, EXEC, Inhouse (MyGenostics), and HGMD.
Selection of potentially pathogenic mutations for downstream analysis was carried out based on five criteria: (i) Mutation with >5 reads and a mutation rate >30% were included; (ii) mutations with frequencies >5% in the 1000 Genomes, ESP6500, and in-house databases were excluded; (iii) mutations present in the InNormal database (MyGenostics) were excluded; (iv) synonymous variants were excluded; (v) after assessing criteria (i), (ii), and (iii), if the mutation was synonymous and had been reported in HGMD, it was included. Any mutations retained after filterings were expected to be pathogenic. Candidate variable sites were confirmed via Sanger sequencing using an ABI 3730 analyzer (Applied Biosystems).
2.2.2 Conservation analysis
This amino acid site was conserved in many species ().
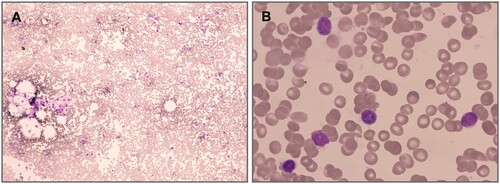
Figure 2. Bone marrow aspiration showed hypo-cellular marrow.
3. Results
3.1 Clinical phenotype
The female patient presented with infusion dependent pancytopenia a few days after birth, and her intellectual and psychomotor developments were behind peers of the same age a few months after birth. The diagnosis of global developmental delay was confirmed after evaluation by the rehabilitation physician. Blood results were as follows: HB 20∼89 g/L, WBC 0.8∼2.7 × 109/L, N% 8∼15, PLT 2∼75 × 109/L, REC% 0.15∼0.25, MCV 83∼89 fL. Bone marrow aspiration revealed: reduced trilineage hematopoiesis and empty bone marrow particles. A bone marrow biopsy showed hypocellular marrow with a percentage of <10% (). Flow cytometric analysis except to other causes of pancytopenia. The methylated DNA was normally methylated. Ultrasonic cardiogram results revealed an atrial septal defect and patent ductus arteriosus.
3.2 Genetic analysis
Next-generation sequencing identified one heterozygous nonsense mutation in exon 17 of the KAT6A gene (NM_006766), c.4177G > T (p.E1393*), which was predicted to cause truncation within the acidic domain of the protein. Genealogical analysis did not detect variation at this locus in either parent (). No correlational studies have been reported on this locus, and no pathogenicity analysis is available for this locus in the ClinVar database, suggesting that this may be a newly identified mutation. Because the mutation is considered a nonsense mutation, no prediction could be made using software such as SIFT or PolyPhen-2 however, predictions made using MutationTaster, LRT, and GERP++ indicated that the mutation was harmful. According to the genetic variant classification and guidelines of the American College of Medical Genetics and Genomics [Citation8], the variant was tentatively determined to be disease-causing (pathogenic) PVS1 + PS2 + PM2 (PVS1: the variant is a nonsense mutation that may lead to loss of gene function; PS2: no variation at this locus in the patient’s father or mother through pedigree verification analysis; suggesting a spontaneous mutation; PM2: the frequency indicated in the normal population database is-, indicating that the variant that occurs at low frequency). Taken together, our findings suggest that p.E1393* is located upstream and disrupts multiple known functional protein domains and is predicted to be pathogenic.
Figure 3. Facial features of the female proband. Lateral view (A) and frontal view (B) showing facial dysmorphism.
4. Discussion
We report the case of a child with KAT6A syndrome who had a de novo mutation, representing a new variant that has not been previously reported. The nonsense mutation occurs in exon 17 and causes proteins to have a truncated C-terminus, which in turn affects their function. Global developmental delay is a clinical presentation in almost all patients with KAT6A syndrome; it occurs early in life and is commonly the most prominent chief complaint. In the present case, the linguistic and intellectual development of the child was behind that of same-age children since before one year of age, and there was no change in muscle tone. These clinical manifestations were all classical clinical phenotypes reported in the literature, further confirming that this newly identified mutation expands the mutational spectrum of KAT6A syndrome. Therefore, studies on the correlation between genotypes and clinical phenotypes are of particular importance.
In this case, the occurrence of pancytopenia was particularly notable. The child’s bone marrow morphology and the biopsy supported the presence of bone marrow failure, a clinical presentation that has not been reported in the literature. Moreover, the patient had pancytopenia since birth. After gene sequencing, common congenital bone marrow failure syndromes, such as Fanconi anaemia were excluded. In fact, it meets the diagnostic criteria for unclassified congenital bone marrow failure syndrome [Citation9]. Hence, more attention should be paid to the correlation between pancytopenia and the KAT6A gene.
As mentioned in the introduction, there are very few reports on KAT6A syndrome involving the hematological system. Although sufficient evidence is lacking, researchers believe that the number of reported cases of the syndrome is currently too small to capture the full range of phenotypes and that further research is needed. Animal studies have found that KAT6A is required for the normal development of the thymus, hematopoietic system, and skeleton in rodents, and is expressed in most tissues in embryonic and adult mice. Mice with homozygous KAT6A loss-of-function mutations fail to develop hematopoietic stem cells during embryogenesis. Surprisingly, hematopoietic progenitor cells developed at an early stage when KAT6A was absent. Although their numbers were reduced, these progenitor cells were able to form all mature hematopoietic cell types. Fetal KAT6A-deficient mice have normal hematocrits despite delayed erythrocyte maturation. These results suggest that KAT6A is essential for hematopoietic stem cell development but less important for the differentiation of progenitor cells [Citation10]. Our patient provides an example of bone marrow failure in humans resulting from a patient with a KAT6A mutation causing a truncated C-terminus. The hypothesis surrounding this pathogenic variant, specifically it affects both hematopoietic stem cell development and the differentiation of progenitor cells, warrants further investigation. The fusion gene t(8;16)(p11;p13) formed by KAT6A and CREBBP has a known role in acute myeloid leukaemia, suggesting a potential relationship between the KAT6A gene and hematologic disorders [Citation11].
The KAT6A gene was found to be expressed in 49 of 79 types of tissues analyzed by the Human Protein Atlas, including the brain (high) and heart (low) [Citation12]. Because it encodes the histone acetylation component of a multiprotein complex, this component is susceptible to genetic, epigenetic, and environmental modifiers. Considering the aforementioned factors, variability or complexity of the clinical phenotypes of patients with KAT6A syndrome is expected. Some researchers have classified KAT6A mutations into two categories, early-onset protein truncation mutations (exons 1–8) and late-onset protein truncation mutations (exons 16–17) based on the site of mutation occurrence, and concluded that those with the latter tend to have more severe intellectual and developmental disabilities and are more likely to have other clinical symptoms [Citation6]. Earlier animal studies also expounded the importance of the local C-terminal domain, where haploinsufficiency as a result of protein truncation due to mutations in this region was more likely to lead to clinical symptoms [Citation12]. The serine/methionine-rich domain of the C-terminus has been shown to possess transcription activation activity [Citation13], for example, KAT6A can interact with RUNX2 through this domain, and special attention should be paid to the importance of RUNX2 for skeletal development [Citation14]. Some studies have reported role of aberrant KAT6A expression in the pathogenesis of some developmental disorders. Trinh et al. described a girl and her father with a heterozygous KAT6A variant (c.5924A > G, p.N1975S) containing a mutation in the C-terminal transactivation domain of the protein a heterozygous variant in the C-terminal transactivation domain of the protein who exhibited milder phenotypes than previous reported patients with truncating mutations [Citation15]. It seems clear that our patient exhibited almost known clinical phenotypes of KAT6A syndrome.
Molecular diagnosis can be a way to shift from phenotype-driven management of clinical symptoms to a more refined treatment based on genotype [Citation15]. Kelley et al. have shown that a considerable proportion of KAT6A patients have mitochondrial dysfunction, and the application of mitochondrial antioxidant cocktail therapy can improve their language and motor skills [Citation16]. Based on the known role of KAT6A in histone acetylation, it is tempting to speculate that drug therapy might restore a more normal acetylation, profile and reduce the developmental delay in individuals with KAT6A mutations. HDAC inhibitors are regarded as potential drugs; however, considering that these drugs may disturb complex signaling pathways, many related studies must be performed before clinical application [Citation17].
The proband in this study inspired a series of thoughts on the pathogenesis of KAT6A syndrome. It will be important to determine whether the levels of histone acetylation and DNA methylation contribute to the clinical phenotype of the disorder via an epigenetic mechanism. Additionally, future work should investigate whether the application of mitochondrial antioxidant cocktail therapy can change epigenetic indicators while improving symptoms. Because traditional drugs used to treat bone marrow failure such as calcineurin inhibitors are likely to be ineffective from the perspective of pathogenesis, it will also be important to determine whether newer drugs such as rapamycin, G-CSF, and TPO can be used to effectively treat KAT6A syndrome.
Unlike Kelley et al. who reported the KAT6A child with pancytopenia and hyper-methylated DNA but did not describe it in detail [Citation16], we did not try HDAC therapy because our patient and normal DNA methylation, so we did not try HDAC therapy. Of the treatments our patient has received to date, PEG-rhG-CSF treatment showed good efficacy. Moving forward, we intend to conduct long-term monitoring of relevant indicators, and we will consider the use of some other methods, such as hematopoietic stem cell transplantation, in our treatment of the patient.
Authors’ contributions
LJ, YC, XY, JY and SC participated in clinical treatment and collected the clinical data. QA interpreted data and wrote the paper.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in the paper.
Ethics approval
The study was approved by the Ethics Committee of the Tianjin Children’s Hospital.
Consent to participate
Informed consent had been obtained from the patient’s legal guardians.
Acknowledgements
We thank Dr. Chunquan Cai and Dr. Jianbo Shu for their treatment recommendations and assistance.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Zhou C, Liu W, Duan Y. MOZ/KAT6A: a promising target for acute myeloid leukemia therapy. Future Sci. 2020;12(9):759–761.
- Yang X-J. MOZ and MORF acetyltransferases: Molecular interaction, animal development and human disease. Biochim Biophys Acta (BBA)-Mol Cell Res. 2015;1853(8):1818–1826.
- Millan F, Cho MT, Retterer K, et al. Whole exome sequencing reveals de novo pathogenic variants in KAT6A as a cause of a neurodevelopmental disorder. Am J Med Genet Part A. 2016;170(7):1791–1798.
- Urreizti R, Lopez-Martin E, Martinez-Monseny A, et al. The challenges of living with and managing epidermolysis bullosa: insights from patients and caregivers. Orphanet J Rare Dis. 2020;15(1):1–14.
- A. Gauthier-Vasserot, C. Thauvin-Robinet, A.L. Bruel, Y. Duffourd, J. St-Onge, T. Jouan, J.B. Rivière, D. Heron, J. Donadieu, C. Bellanné-Chantelot, Application of whole-exome sequencing to unravel the molecular basis of undiagnosed syndromic congenital neutropenia with intellectual disability, Am J Med Genet Part A. 2017;173(1:62–71.
- Kennedy J, Goudie D, Blair E, et al. KAT6A syndrome: genotype–phenotype correlation in 76 patients with pathogenic KAT6A variants. Genet Med. 2019;21(4):850–860.
- Smith C, Harris J. Sleep, behavior, and adaptive function in KAT6A syndrome. Brain Sci. 2021;11(8):966.
- Li Q, Wang K. InterVar: clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am J Human Genet. 2017;100(2):267–280.
- Teo JT, Klaassen R, Fernandez CV, et al. Clinical and genetic analysis of unclassifiable inherited bone marrow failure syndromes. Pediatrics. 2008;122(1):e139–e148.
- Sheikh BN, Yang Y, Schreuder J, et al. MOZ (KAT6A) is essential for the maintenance of classically defined adult hematopoietic stem cells. Blood. J Am Soc Hematol. 2016;128(19):2307–2318.
- Xie W, Hu S, Xu J, et al. Acute myeloid leukemia with t(8;16)(p11.2;p13.3)/KAT6A-CREBBP in adults; 16)(p11. 2; p13. 3)/KAT6A-CREBBP in Adults. Ann Hematol. 2019;98(5):1149–1157.
- Tham E, Lindstrand A, Santani A, et al. Dominant mutations in KAT6A cause intellectual disability with recognizable syndromic features. Am J Human Genet. 2015;96(3):507–513.
- Champagne N, Bertos NR, Pelletier N, et al. Identification of a human histone acetyltransferase related to monocytic leukemia zinc finger protein. J Biol Chem. 1999;274(40):28528–28536.
- Pelletier N, Champagne N, Stifani S, et al. MOZ and MORF histone acetyltransferases interact with the Runt-domain transcription factor Runx2. Oncogene. 2002;21(17):2729–2740.
- Trinh J, Hüning I, Yüksel Z, et al. A KAT6A variant in a family with autosomal dominantly inherited microcephaly and developmental delay. J Hum Genet. 2018;63(9):997–1001.
- Kelley RI. KAT6A syndrome: deficiency of a histone acetyltransferase as the cause of mild to severe mitochondrial disease. Am J Med Genet Part A. 2019;179(4):729–730.
- Arboleda VA, Lee H, Dorrani N, et al. De novo nonsense mutations in KAT6A, a lysine acetyl-transferase gene, cause a syndrome including microcephaly and global developmental delay. Am J Human Genet. 2015;96(3):498–506.