
ABSTRACT
Objectives:
To report the long-term prophylaxis management of a child with type 3 von Willebrand disease by switching to Wilate (Octapharma AG), a plasma-derived, double virus-inactivated concentrate of freeze-dried of a 1 to 1 ratio of active Von Willebrand Factor and Factor VIII (pdVWF:pdFVIII) recently marketed as Eqwilate in France.
Methods:
This is a case report of 12.6-year-old boy with congenital Type 3 VWD who had a history of frequent bleeds. Prophylaxis started at the age of 38 months with FVIII-poor pdVWF concentrate (Wilfactin, LFB) and FVIII (Wilstart, LFB). Pharmacokinetics and thrombin generation assay were performed. Annualized bleeding rate was derived from the bleeding episodes documented in the medical record during a 24-month period before and after starting pdVWF:pdFVIII concentrate.
Results:
Both product injections promptly raised the endogenous thrombin potential (ETP). However, the maximal concentration of formed thrombin was higher following pdVWF:pdFVIII injection. Due to a high bleeds frequency and better results regarding FVIII levels and thrombin generation, the prophylaxis regimen was changed to the same dose and frequency of pdVWF:pdFVIII concentrate (42 IU/kg per day, three times a week). During the last 24 months, annualized total, trauma, and spontaneous bleeding rates were 7.5, 4.5, and 3, respectively. These rates decreased to 2, 1.5, and 0.5 respectively during the next two years. The mother reported a marked improvement in the quality of life of his son and hers.
Conclusion:
Switch to pdVWF:pdFVIII concentrate for long-term prophylaxis in a young type 3 VWD patient was safe and effective in reducing bleeds.
Introduction
Von Willebrand disease (VWD) is the most common hereditary bleeding disorder. The current International Society on Thrombosis and Haemostasis (ISTH) classification recognizes three types: type 1 is a partial quantitative deficiency of von Willebrand factor (VWF), type 2 is caused by qualitative abnormalities of VWF, and type 3 is a virtual absence of the VWF protein with associated deficient Factor VIII (FVIII) levels [Citation1]. Mucocutaneous bleedings (epistaxis, menorrhagia, easy bruising, prolonged bleeding from minor wounds and the oral cavity, and gastrointestinal bleeding) are typical of VWD [Citation2]. Patients with VWD are usually managed through ‘on-demand’ treatment by increasing or supplementing missing VWF at the onset of bleeding [Citation3] or as short-term perioperative prophylaxis in patients with VWD undergoing surgery [Citation4,Citation5]. Recent guidelines recommend long-term prophylaxis in patients with VWD and a history of severe and frequent bleeds, i.e. long-term secondary prophylaxis [Citation6]. Definition of long-term may vary between studies and guidelines, but 6 months minimum is typically what can be considered [Citation6]. Long-term prophylaxis has thus been reported to be effective [Citation7,Citation8] and a group of European experts in VWD highlighted the place of secondary long-term prophylaxis in the management of severe forms of VWD [Citation9]. A summary of evidence performed by Connell et al [Citation6] reported notably the reduced risk of bleeding episodes (rate ratio [RR], 0.24; 95% confidence interval [CI], 0.17–0.35) in the only randomized controlled trial they found [Citation10]. Based on other evidence from observational studies, the authors also reported a reduced risk of bleeding episodes (RR, 0.34; 95% CI, 0.25–0.46) and hospitalization (RR, 0.64; 95% CI, 0.44–0.93), confirming the interest of this treatment strategy, although they graded the level of evidence as low [Citation6]. Wilate (Octapharma AG, Lachen, Switzerland) is a plasma-derived, double virus-inactivated concentrate of freeze-dried of active VWF and FVIII (pdVWF:pdFVIII) in a 1:1 ratio. It was recently marketed in France under the name Eqwilate. At the same time, it was already available in several countries worldwide. We report here the switch to the pdVWF:pdFVIII concentrate for the prophylaxis in a paediatric patient with Type 3 VWD who was hardly controlled with his previous regimen based on FVIII-poor pdVWF concentrate as illustrated with the frequent bleeding episodes (epistaxis, trauma-related hematomas) that required on-demand treatment of FVIII-poor pdVWF.
Ethics
Written informed consent was obtained from the parent to publish the patient’s details.
Case
History
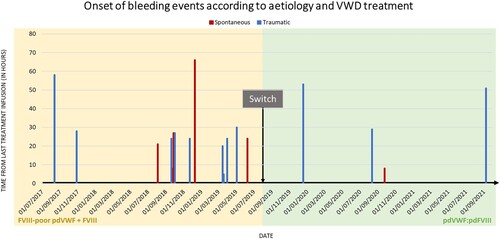
The patient was a 12.6-year-old boy with congenital type 3 VWD with VWF antigen and activity levels of 7% and 4%, respectively, and FVIII:C% levels of 21%. The heterozygous mutations were Exon 45 c.7603C > T (p.Arg2535X) and Exon 53 c.8280C > G (p.Tyr2760X). He had asthma and a food allergy. Before prophylaxis initiation, he suffered from left and right ankle hemarthrosis and right and left knee hemarthrosis within the same year. Prophylaxis started at the age of 38 months with 1000 IU FVIII-poor pdVWF concentrate (Wilfactin, LFB, Les Ulis, France) and 500 IU FVIII (Factane, LFB, Les Ulis, France) every week and rapidly increased to 1000 IU FVIII-poor pdVWF two times a week with the installation of an implantable port. Eventually, the prophylaxis regimen was increased to 2000 IU FVIII-poor pdVWF three times a week after three years. Despite maintenance of long term-prophylaxis, he experienced frequent bleeding episodes requiring on-demand treatment with FVIII-poor pdVWF as illustrated during the last 24 months before the switch to new therapy (). Annualized total, trauma, and spontaneous bleeding rates were 7.5, 4.5, and 3, respectively (excluding bleedings from tooth exfoliation). No inhibitory anti-FVIII antibody nor anti-VWF antibody was developed.
Figure 1. The onset of bleeding events according to aetiology and VWD treatment. The time from the last treatment infusion at spontaneous bleeding onset varied from 8 and 66 h during the pdVWF:pdFVIII period. The start of spontaneous bleeding occurred 8 h after the last treatment infusion during the FVIII-poor pdVWF period. Time from last treatment infusion at spontaneous bleeding onset varied from 5 and 58 h during the pdVWF:pdFVIII period and between 29 and 53 h during the FVIII-poor pdVWF period. Abbreviations: FVIII, factor VIII; pd, plasma-derived; VWD, von Willebrand disease; VWF, von Willebrand factor.
Pharmacokinetics and thrombin generation time
Following the recent marketing authorization in France of the pdVWF:pdFVIII concentrate, we performed a pharmacokinetic (PK) analysis with a time interval of six days after one with the current treatment.
Overall, FVIII-poor pdVWF concentrate and pdVWF:pdFVIII concentrate infusions had similar effects on VWF activity over time but showed distinct profiles of FVIII activity. Numerically, the peak VWF:Act at 30 min was slightly higher with pdVWF:pdFVIII concentrate (68%) vs. the one with FVIII-poor pdVWF concentrate (61%) (). This was associated with a higher recovery of FVIII activity levels, which was expected as no FVIII is present in FVIII-poor pdVWF concentrate.
Figure 2 Pharmacokinetics analysis of pdVWF:pdFVIII and FVIII-poor pdVWF concentrates and the peak of thrombin generation (after infusion 42 IU/kg VWF) from platelet-poor plasma. Left axis: Peak of thrombin generation (concentration in nM), normal range (5-95th percentiles) 53–115 nM. Right axis: VWF or FVIII activity in percentage; VWF:Act% was measured using the VWF gain-of-function glycoprotein Ib assay (Innovance VWF Ac assay, Siemens), normal range for O blood type 124–49%. FVIII:C% was measured by the chromogenic assay (Biophen FVIII:C, Hyphen Biomed), normal range 70–150%. Abbreviations: FVIII, factor VIII; pd, plasma-derived; PPP; platelet-poor plasma; VWF, von Willebrand factor, Act%, activity percentage: C%, procoagulant activity percentage.
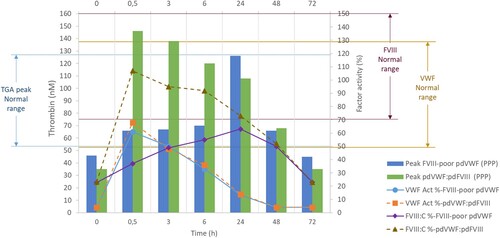
A thrombin generation assay (TGA) was performed on platelet-poor plasma from the PK analysis samples. Both product injections raised the endogenous thrombin potential (ETP) promptly, as soon as 30 min, to higher ranges of average values from healthy volunteers or slightly above ().
Table 1. Thrombin generation assay on PPP pharmacokinetics samples after pdVWF:pdFVIII and FVIII-poor pdVWF concentrates administration (after infusion 42 IU/kg VWF).
However, the maximal concentration of formed thrombin (peak) was higher following pdVWF:pdFVIII injection. This was associated with a velocity that reached average values from healthy volunteers, unlike those observed after FVIII-poor pdVWF concentrate injection (except at 24 h).
Treatment plan
Due to a high number of bleeds per year and in view of the results showing better factor VIII levels and thrombin generation, we chose to switch to pdVWF:pdFVIII concentrate (42 IU/kg) per day, three times a week.
Outcomes
After four months, no complementary injection was required. The patient did not suffer from gum bleedings, petechiae, or epistaxis. The percutaneously implantable chamber had to be removed due to a Staphylococcus infection treated without complications. However, it was not replaced as it was no longer required based on the recent bleeding profile.
The prophylaxis regimen was adjusted to 42 IU/kg per day, twice a week, and continued without interruption.
Twenty-four months after the switch, three trauma (ankle sprain, right finger hemarthrosis, and left ankle hemarthrosis) and one spontaneous bleeding (left shoulder pain) occurred, as retrospectively collected from the medical records (). This corresponded to an annualized total, trauma, and spontaneous bleeding rates of 2, 1.5, and 0.5, respectively. Mean (SD) time between last infusion and onset of bleedings was, respectively, 27.2 ± 15.7 h and 35.1 ± 21.2 h. Compared to the period with the previous regimen during which 60% (9/15) of the bleeding episodes occurred within 24 h from last infusion, only 25% (1/4) bleeding episodes occurred within this timeframe (p = 0.3034, Fisher’s exact test).
No adverse reaction was reported.
The mother reported a marked improvement in the quality of life of his son and hers.
Discussion
Initiation and maintenance of long-term prophylaxis for patients with VWD are not as standard as for patients with haemophilia, even for patients with Type 3 VWD. Although prophylaxis is being proposed to an increased number of patients with VWD, the consensus about indications among experts is lacking [Citation11]. The corpus of published data, notably in real-life settings, is limited. An important question is the planning of prophylaxis concerning the composition of the VWF concentrates, i.e. with or without FVIII [Citation11]. Recently, an observational post-marketing study investigated the effectiveness and safety of pdVWF:pdFVIII concentrate in a cohort of patients with VWD, among which prophylaxis involved 25 out of the 111 patients. During the 6-month period prior to entering the study, a median annualized bleeding rate of 12 (range 0–208) was reported for these patients. The prophylaxis with pdVWF:pdFVIII concentrate was then associated with a marked improvement (6-fold decrease) in the annualized bleeding rate with a median value of 1.9 bleeding episodes per year [Citation12]. Here, switching to pdVWF:pdFVIII concentrate resulted in an essential reduction in bleeding episodes of similar magnitude. This was also in line with other studies involving other concentrates as suggested by the recent summary of evidence performed by Connell et al [Citation6]. The authors reported a relative risk of bleeding episodes ranging from 0.24 (95% CI, 0.17–0.35) to 0.34 (95% CI, 0.25–0.46), depending on whether data originated, respectively, from a single randomized-controlled trial (n = 19) or 5 pre–post observational studies with explicit comparative data [Citation6]. Consistently, the absolute annualized bleeding rate for total bleeding episodes derived from the 24-month of prophylaxis with the pdVWF:pdFVIII concentrate was within the range of the reported pooled rate of 3.20 (95% CI, 1.96–5.24) bleeding episodes per patient per year, using data from observational studies without explicit comparative data [Citation6]. Furthermore, most of the bleeding episodes occurred after 24 h from last infusion (unlike those during the period with the previous regimen). Although not statistically significant (likely due to obvious lack of power), this may suggest that the first 24 h post infusion were less prone to bleedings, probably in relation with the pharmacokinetics profile of the pdVWF:pdFVIII concentrate. This resulted in a substantial change in the daily life of the family. The implantable port was no longer judged required, alleviating part of the prolonged and repeated IV administration burden.
The TGA is a global assay of haemostasis reflecting the overall functional state of the clotting system. Its use as diagnosis or bleeding risk assessment is debated [Citation13]. Nonetheless, in patients with VWD, Rugeri et al. reported that a low thrombin peak was associated with higher bleeding risk [Citation14]. Furthermore, the authors showed that in VWD type 3 patients, TGA parameters were mainly dependent on FVIII rather than VWF. Here, higher peak, velocity, and ETP following pdVWF:pdFVIII than FVIII-poor pdVWF illustrated a rapid capacity to generate thrombin supporting the role of FVIII in this process. This particular PK and TGA characteristic conferred by the concomitant administration of VWF and FVIII may translate more stably into a less severe VWD phenotype.
Conclusion
Switch from FVIII-poor pdVWF to pdVWF:pdFVIII concentrate for long-term prophylaxis in a young type 3 VWD patient was safe and effective in reducing the number of bleeding episodes.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Sadler JE, Budde U, Eikenboom JCJ, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand factor. J Thromb Haemost. 2006;4(10):2103–2114.
- Sadler JE, Mannucci PM, Berntorp E, et al. Impact, diagnosis and treatment of von Willebrand disease. Thromb Haemost. 2000;84(2):160–174.
- Ingerslev J, Hvitfeldt Poulsen L, Sorensen B. Current treatment of von Willebrand’s disease. Hamostaseologie. 2004;24(1):56–64.
- Federici AB. Management of von Willebrand disease with factor VIII/von Willebrand factor concentrates: results from current studies and surveys. Blood Coagul Fibrinolysis. 2005;16(Suppl 1):S17–S21.
- Nichols WL, Hultin MB, James AH, et al. Von Willebrand disease (VWD): evidence-based diagnosis and management guidelines, the National Heart, Lung, and Blood Institute (NHLBI) Expert Panel report (USA). Haemophilia. 2008;14(2):171–232.
- Connell NT, Flood VH, Brignardello-Petersen R, et al. ASH ISTH NHF WFH 2021 guidelines on the management of von Willebrand disease. Blood Advances. 2021;5(1):301–325.
- Berntorp E, Petrini P. Long-term prophylaxis in von Willebrand disease. Blood Coagul Fibrinolysis. 2005;16(Suppl 1):S23–S26.
- Federici AB, Barillari G, Zanon E, et al. Efficacy and safety of highly purified, doubly virus-inactivated VWF/FVIII concentrates in inherited von Willebrand’s disease: results of an Italian cohort study on 120 patients characterized by bleeding severity score. Haemophilia. 2010;16(1):101–110.
- Castaman G, Goodeve A, Eikenboom J. Principles of care for the diagnosis and treatment of von Willebrand disease. Haematologica. 2013;98(5):667–674.
- Peyvandi F, et al. A phase III study comparing secondary long-term prophylaxis versus on-demand treatment with vWF/FVIII concentrates in severe inherited von Willebrand disease. Blood Transfus. 2019;17(5):391–398.
- Federici AB. Prophylaxis in patients with von Willebrand disease: who, when, how? J Thromb Haemost. 2015;13(9):1581–1584.
- Sholzberg M, Khair K, Yaish H, et al. Real-world data on the effectiveness and safety of wilate for the treatment of von Willebrand disease. TH Open. 2021;5(3):e264–e272.
- Holm E, Zetterberg E, Lövdahl S, et al. Patients referred for bleeding symptoms of unknown cause: does evaluation of Thrombin generation contribute to diagnosis? Mediterr J Hematol Infect Dis. 2016;8(1):e2016014.
- Rugeri L, Beguin S, Hemker C, et al. Thrombin-generating capacity in patients with von willebrand's disease. Haematologica. 2007;92(12):1639–1646.