
ABSTRACT
Background
Hb Chapel Hill [Alpha2 74(EF3) Asp > Gly] results from an GAC > GGC substitution at codon 74 of the HBA1 or HBA2 genes. Hb Chapel Hill has not been reported since 1986.
Methods
A heterozygous mutation, HBA2: c.224A > G, was identified in the proband, her father and sister. We compared the haematological and clinical data of this family with the data reported in the limited number of individuals.
Results
Having excluded iron deficiency, the Hb Chapel Hill was asymptomatic in heterozygous state. The cases presented here characterize cases in new techniques including capillary electrophoresis (CE). Two aberrant peaks were identified by CE, a major peak migrating in the zone 7 that correspond to Hb Chapel Hill (αChapel Hill 2β2) and a minor peak migrating in the zone 1 that correspond to Hb Chapel Hill2 (αChapel Hill 2δ2). Focusing on the variant expression, the Hb Chapel Hill plus Hb A2 variant were around 18.9–20.6% of total Hb in three members.
Conclusion
This data will be useful for providing up-to-date and high quality information on the Hb Chapel Hill.
Introduction
In 21 regions of Guangdong Province, 16.83% of couples (pregnant women and their husbands) are heterozygous carriers of α-or β-thalassemia [Citation1]. These more severe thalassemia phenotypes are mostly observed in populations where common thalassemia determinants are more frequent [Citation2,Citation3]. For some hemoglobin variants, carriers are asymptomatic with normal or nearly normal Hb levels, whereas non-deletional HbH disease manifests as hydrops fetalis or α-thalassemia intermedia [Citation4,Citation5]. 156 unstable hemoglobins are listed in the hemoglobin variant database (https://globin.bx.psu.edu/cgi-bin/hbvar/counter). The four most frequent Hb variants in southern China were Hb E, Hb New York (mildly unstable), Hb J-Bangkok, and Hb Q-Thailand [Citation6]. Hb Chapel Hill is a rare mildly unstable hemoglobin characterized by the A > G substitution at position 224 in the HBA2 of the α-globin gene [Citation7]. It was difficult to completely rule out all Hb variants with the existing screening methods. As the rare variants have rarely been reported before, thus making it difficult to get more data. Future work improves the quality and accuracy of the data.
Few cases of Hb Chapel Hill (HBA2: c.224 A > G) are known. It was a type of mildly unstable Hb who was first reported in a 45-year-old Caucasian female living in the city of Chapel Hill which was located in North Carolina in 1976 [Citation8]. The second observation was found in a 42-year-old female with a β-thalassemia trait from Guangxi Zhuang Autonomous Region of southern China in 1986 [Citation9]. However, after around four decades, we have seen progress in the techniques used to identify, there is no debate about Hb Chapel Hill update. As this mutation was found in a limited number of cases, the characterization of Hb Chapel Hill was not sufficiently described. Herein, we describe a rare mutation in a Chinese family from Foshan City, Guangdong province of southern China. Further, no other published reports used the same variant identification methodology we describe here.
Materials and methods
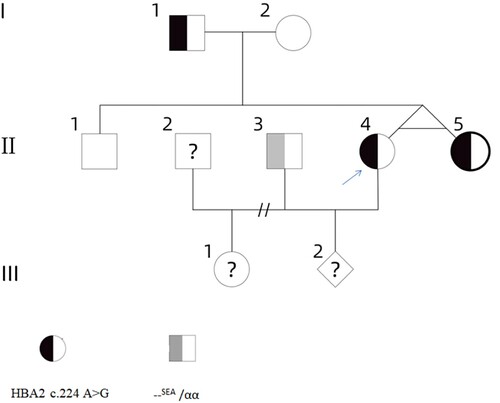
A 33-year-old women was referred to our hospital because of the observation of an abnormal Hb fraction detected by capillary electrophoresis in another hospital. The abnormal Hb fraction was more carefully investigated when the patient was pregnant with her second child at 15 weeks’ gestation. After obtaining informed consent from the family members, five members of a three-generation family were examined (). This study was approved by the Medical Ethics Committee of the Guangdong Women and Children Hospital (NO.202201168).
Figure 1. The family pedigree: the proband (II-4) is marked with an arrow.
Hematological parameters were determined by an automatic blood cell analyzer (BC-7500cs, Mairui, Shenzhen, China). MCV <82 fL and/or MCH <27 pg was suspected as thalassemia carriers. In such cases, having excluded iron deficiency anemia, Hb analysis was conducted using automatic Hb capillary electrophoresis (CE) apparatus (CapillaryS2, Sebia, Lisses, France). Genomic DNA was obtained from peripheral venous blood using an automation nucleic acid extractor Lab-Aid 820 (Zee San Biotech Company, Fujian, China). The suspension-array system (DaAn Gene Company, Guangzhou, China) which had been patented in the People’s Republic of China was used to detect the three common deletion α-thalassemia(-α3.7, -α4.2, –SEA), three common non-deletion α-thalassemia (Hb ConstantSpring, Hb QuongSze and Hb Westmead) and 17 common mutations associated with β-thalassemia in the Chinese population [Citation10].
The unknown α-globin gene mutation was analyzed by direct nucleotide sequencing. The primers were as follows: α1-F: 5'-TGGAGGGTGGAGACGT
CCTG-3’; α1-R: 5'-TCCATCCCCTCCTCCCGCCCCTGCC TTTTC-3',α2-F: 5'-GATGGGCGGGAGTGGAGT-3; α2-R: 5'-GGACAGGGGATGGTTCAGC-3’. The cycling conditions involved initial denaturation at 95°C for 5 min followed by 33 cycles of denaturation at 95°C for 40 s, annealing at 66°C for 30 s with an extension at 72°C for 70 s with a final extension at 72°C for 7 min.
Results
The haematological data of the family are shown in . Three of them presented with abnormal Hb fraction (). Hb X moves to the position of zone 7, the falsely increased levels of Hb F may result from the migration of abnormal hemoglobin to Hb F region. Although the thalassemia screening is positive, her thalassemia genotype results are normal using the routine kits. The DNA analysis of the proband (II-4) characterized a rare mutations on the α2-globin gene: the GAC > GGC substitution at codon 74, predicted to change the Asp to a Gly at position 75 of the α2 chain, thus resulting in Chapel Hill. The gene analysis in the proband, her father (I-1) and sister (II-5) revealed that they carry the same gene variant (), whereas her mother (I-2) and brother(II-1) are normal individuals. The proband and her sister are identical twins which are confirmed by subsequent linkage analysis of their DNA with short tandem repeat markers. Her husband (II-3) is reported to be –SEA /αα in another hospital, but no hematological data is available. Due to the II-2 is the ex-husband of the proband, which neither make the study of II-2 impossible nor does their child (III-1).
Figure 2. Electropherograms of hemoglobin A plus hemoglobin Chapel Hill, the peaks from left to right being Hb A, Hb Chapel Hill (αChapel Hill 2β2) (zone 7), Hb A2, and Hb Chapel Hill2 (αChapel Hill 2δ2) (zone 1).
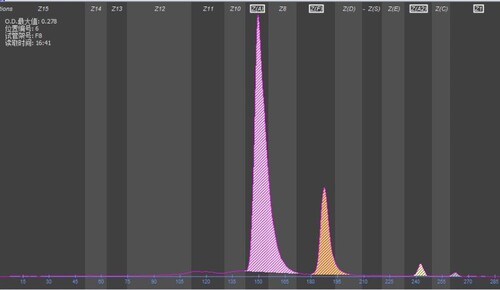
Figure 3. DNA sequencing analysis showing the mutation of HBA2: c.224A > G on the α2 gene. (arrow indicates heterozygous HBA2: c. 224A > G mutation).
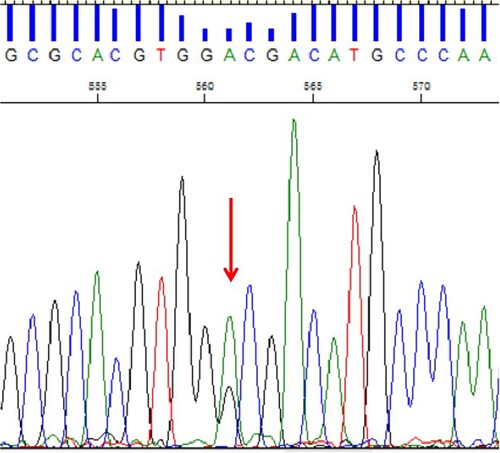
Table 1. Hematological data and genotypes of the family members.
Discussion
With other mutation and other factor affecting the expression or the percentage of Hb Chapel Hill was usual. After reviewing the literature, the first report where they unexpectedly found a patient to have HB Chapel Hill, while evaluating a woman for erythrocytosis [Citation8]. The second case was found in a family in association with β-Thalassemia [Citation9]. The cases presented here were ideal samples to be characterized in alternative methodology including capillary electrophoresis (CE). During assessment of Hb Chapel Hill expression in this study, we noticed that the Hb X quantity was stable, ranging from 18.2% to 20.4% of total Hb on CE. Two aberrant peaks were identified in proband, a major peak migrating in the Hb F zone constituting 18.4% and a minor peak migrating in zone 1 (0.7%) (). DNA sequencing strategy for α globin gene was recommended when a major peak (α2Xβ2) present with a corresponding Hb A2 variant peak (α2Xδ2). Hb A2 is a hemoglobin tetramer composed of two α and two β-globin chains (α2δ2). When an α-globin chain variant (αX) is present, HbA2 would be present at a low percentage of the total Hb [Citation11–13].
The mutation was inherited from the proband’s father (I-1) and was also present in proband’s sister (II-5). The hematological parameters of the father and sister in present case showed some similarity with the first report in which normal levels of MCV and MCH were also present [Citation8]. However, the relatively low levels of ferritin detected in both proband (4.71 ng/mL) and her sister (4.91 ng/mL) (reference range 4.63–204 ng/mL). It was noted that her sister showed no anemia (14.6 g/dL), may be attributed to the iron deficiency anemia which was a dynamic process of development. Considering pregnancy, these results indicated that the proband may had iron deficiency anemia (12.2 g/dL), which may lead to the decreased levels of MCV and MCH. Having excluded iron deficiency, the proband’s father did not present with any clinical symptoms or hematological changes related to the variant. Based on hematological and clinical evaluations, it could be expected that the rare mutation HBA2: c.224 A > G excluding iron deficiency anemia would result in normal levels of MCV and no clinical significance in heterozygous state.
All reported Hb Chapel Hil cases were located in α2 of the α-globin gene complex. However, the case had not been reported compound heterozygous for Hb Chapel Hill in α2 gene and deletional form α thalassemia. The children of the proband were unavailable for study, therefore we were not able to verify whether the children of the proband (III-1 and III-2) were carriers or not. It was noteworthy that the fetus (III-2) had a 25% chance of being non-deletional HbH disease (–SEA /αChapel Hillα). The Asp residue at position 75 of the α2 chain was a conserved residue in globin evolution base on bioinformatic analysis. We speculated that the substitution of a conserved residue was responsible for the instability of the Hb molecule [Citation14]. As the phenotypes of these genotype combinations (--SEA /αChapel Hillα) had not been reported before, thus creating counseling problems. We assumed that the fetus would be α-thalassemia intermedia when the mutation combines with an α deletion defect. We counseled about the risk in their pregnancies and offered prenatal diagnosis. The couples declined the prenatal diagnosis. According to prenatal ultrasound and echocardiographic examination, subsequent development of this fetus is not anomalous till now.
Conclusion
Hb Chapel Hill is mildly unstable but carriers typically have normal clinical pre-sentation and hematological profile. It is now 36 years since the last literature were published concerning Hb Chapel Hill. This study found the rare mutation in the Chinese population, characterized a case in new analytical developments.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Yin A, Li B, Luo M, et al. The prevalence and molecular spectrum of α- and β-globin gene mutations in 14,332 families of Guangdong Province, China. PLoS One. 2014 27;9(2):e89855.
- Shwe S, Boo NY, Ong HK, et al. Haemoglobin Constant Spring (HbA2: c.427T > C) and haemoglobin Adana (HbA2: c.179G > A) in jaundiced Malaysian term neonates with clinically significant hyperbilirubinemia. Malays J Pathol. 2020;42(2):253–257.
- Shang X, Peng Z, Ye Y, et al. Rapid targeted next-generation sequencing platform for molecular screening and clinical genotyping in subjects with hemoglobinopathies. EbioMedicine. 2017;23:150–159.
- Tampaki A, Theodoridou S, Apostolou C, et al. Α case of late diagnosis of compound heterozygosity for Hb Adana (HBA2:c.179G > A) in trans to an α+- thalassemia deletion: guilty or innocent. Hippokratia. 2020;24(1):43–45.
- Singh SA, Sarangi S, Appiah-Kubi A, et al. Hb Adana (HBA2 or HBA1: c.179G > A) and alpha thalassemia: genotype-phenotype correlation. Pediatr Blood Cancer. 2018;65(9):e27220.
- Xu A, Chen W, Xie W, et al. Hemoglobin variants in southern China: results obtained during the measurement of glycated hemoglobin in a large population. Clin Chem Lab Med. 2020;59(1):227–232.
- Giardine BM, Joly P, Pissard S, et al. Clinically relevant updates of the HbVar database of human hemoglobin variants and thalassemia mutations. Nucleic Acids Res. 2021;49(D1):D1192–D1196.
- Orringer EP, Wilson JB, Huisman TH. Hemoglobin Chapel Hill or alpha2 74 Asp replaced by Gly beta2. FEBS Lett. 1976;65(3):297–300.
- Hsu L, Lung QF, Tang ZN, et al. Hb Chapel Hill or alpha 274(EF3)Asp—-Gly beta 2 observed in a Chinese family in association with beta-thalassemia. Hemoglobin. 1986;10(1):77–86.
- Luo MY, Hu TT, Wang JC, et al. Application of a suspension array based on barcoded magnetic beads in thalassemia gene diagnosis. J Trop Med. 2016;16(7):832–835.
- Borbely N, Phelan L, Szydlo R, et al. Capillary zone electrophoresis for haemoglobinopathy diagnosis. J Clin Pathol. 2013;66(1):29–39.
- Du L, Qin D, Wang J, et al. The first Chinese case of unstable hemoglobin Santa Ana detected by capillary electrophoresis: a case report and literature review. Hematology. 2022;27(1):258–262.
- Stephens AD, Angastiniotis M, Baysal E, et al. ICSH recommendations for the measurement of haemoglobin A2. Int J Lab Hematol. 2012;34(1):1–13.
- Wajcman H, Traeger-Synodinos J, Papassotiriou I, et al. Unstable and thalassemic alpha chain hemoglobin variants: a cause of Hb H disease and thalassemia intermedia. Hemoglobin. 2008;32(4):327–349.