
ABSTRACT
Objective
To investigate the molecular diagnosis of hemoglobin variants in Z region by Capillary electrophoresis in Central Guangxi, Southern China, and analyze their distribution and phenotypic characteristics, to provide a reference for clinical consultation and prenatal diagnosis for couples.
Methods
A total of 23,709 subjects were collected for blood routine analysis, hemoglobin analysis, and common α- and β-globin gene loci in Chinese population. The hemoglobin electrophoresis components were divided into Zone 1-Zone 15 (Z1-Z15) by Capillary zone electrophoresis (CE). For samples not clearly detected by the conventional technology, Sanger sequencing, and multiplex ligation-dependent probe amplification (MLPA) were used. Single-molecule real-time (SMRT) sequencing technology was used to analyze rare-type genes in a sample with a structural variation.
Results
Ten rare hemoglobin variants distributed in Z region were detected in 23,709 samples, including Hb Cibeles, which was reported for the first time in Asia; Hb J-Broussais, Hb G-Honolulu and J-Wenchang-Wuming, they were first reported in Guangxi; 1 case of Hb Anti-Lepore Liuzhou, which was a newly discovered hemoglobin variant; hemoglobin variants Hb G-Siriraj, Hb Handsworth, Hb Q-Thailand, Hb Ube-2, Hb NewYork were also detected.
Conclusion
There are a few studies on rare hemoglobin variants in Z region in Southern China. Ten rare hemoglobin variants were found in this study. The hematological phenotype and component content of hemoglobin variants are related to the occurrence of thalassemia. This study enriched the data of rare hemoglobin variants in Southern China and provided a comprehensive data basis for prenatal diagnosis of hemoglobin variants in this area.
1. Introduction
As one of the most common genetic diseases, hemoglobin diseases are mainly divided into two categories: abnormal hemoglobinopathy and thalassemia. An abnormal hemoglobinopathy is a group of hereditary diseases in which the globin gene is defective and the primary structure of the hemoglobin molecule is changed. Some hemoglobin variants may have a thalassemia-like phenotype, which may lead to severe phenotype in patients when they are co-inherited with thalassemia. With the increase of age, some asymptomatic carriers of hemoglobin variants may have gradually aggravated clinical symptoms [Citation1]. More than 700 types of hemoglobin variants have been found in the HbVar database. About 30% of carriers have abnormal hemoglobin function [Citation2]. These variants are obvious regional or group specificity, and different ethnic groups have their own unique gene mutation spectrum [Citation3]. Hemoglobinopathies are mainly distributed in tropical and subtropical regions, with high incidence in Guangdong, Guangxi, Hainan and other provinces south of the Yangtze River in China [Citation4]. Studies over the years have shown that Guangxi is a provincial-level administrative region with the highest carrying rate of hemoglobinopathy in China, and there are many kinds of hemoglobin variants in this region [Citation5]. At present, blood cells and hemoglobin tests are mainly used as a first-tier screening method in China. Methods for the hemoglobin components analysis include high-performance liquid chromatography (HPLC) and automatic capillary electrophoresis (CE) [Citation6]. Among them, CE can detect various variants (S, C, E, D and Hb Bart’s) [Citation7]. It can completely separate the hemoglobin components that cannot be separated by HPLC and has the advantages of high sensitivity and high detection flux. Molecular detection techniques for hemoglobinopathy mainly include Gap polymerase chain reaction (Gap-PCR) [Citation8], reverse dot blot technique (RDB) [Citation9], multiplex ligation-dependent probe amplification (MLPA) [Citation10], and Sanger sequencing [Citation11]. Among them, Gap-PCR and RDB are the first-line detection technologies in China, but the detection range is very limited, and many rare thalassemia genotypes cannot be determined. Sequencing technology is the gold standard for the diagnosis of thalassemia. Compared with traditional methods, the third-generation sequencing technology (SMRT) [Citation12] developed in recent years can detect more hemoglobin variation sites on HBA1, HBA2 and HBB genes, and can resolve the problem that is difficult to be detected by other sequencing technologies. DNA sequencing technology (especially SMRT) has a high cost and it is difficult to popularize temporarily. It can be seen that different techniques have their own advantages and limitations, and there may be missed diagnoses when they are used alone. Although the prevalence of thalassemia in Southern China has been reported by several investigations, there is still no comprehensive analysis of hemoglobin variants using capillary electrophoresis and molecular techniques. In this study, we analyzed the hematological phenotype, component content and thalassemia involved with hemoglobin variants in central Southern China, which enriched the data of hemoglobin variants in this region and provided a comprehensive data basis for the population with hemoglobin variants.
2. Objects and methods
2.1. Object
A total of 23,709 subjects whowent to Liuzhou Maternal and Child Healthcare Hospital in Guangxi, China from 2020 to 2021, included 11,833 males and 11,876 females, ranging in age from 10 days to 84 years old. The average age was 27.59 ± 13.20 years. Three tubes of EDTA-K2 anticoagulant blood (2 mL in each tube) were collected for routine blood analysis, hemoglobin electrophoresis analysis, and thalassemia gene detection. This study was approved by the Ethics Committee of our hospital, and all subjects signed informed consent. This study complies with the requirements of the Declaration of Helsinki.
2.2. Reagents and instruments
XE-2100 blood cell analyzer and supporting reagents from Sysmex Company, Japan; Capillarys2 capillary electrophoresis instrument and supporting reagents from SEBIA Company, France; Lab-Aid 820 automatic nucleic acid extractor and supporting reagents from Xiamen Zhishan Company, China; ABI ProFlex 96 × 2 well amplification instrument and 3500Dx genetic analyzer from ABI Company, United States; Gel Doc gel imager from BIO-RAD, United States; common thalassemia gene detection kits from Shenzhen Yishengtang Biological Enterprise Co., Ltd., China and Shenzhen Yaneng Biological Co., Ltd., China.
2.3. Methods
2.3.1. Hematology screening
We used an XE-2100 blood cell analyzer for blood routine analysis. Hemoglobin electrophoresis was analyzed by the Capillarys2 capillary electrophoresis apparatus. The capillarys system uses liquid electrophoresis in the capillary to separate various hemoglobin and hemoglobin variants in the alkaline buffer with specific PH values by using the mobility of charged molecules and quantifies them at the extreme negative absorption rate of 415 nm. The main analysis indexes and reference ranges are as follows: Hb A > 94.5%; Hb A2, 2.4-3.4%; Hb F < 2.0%. According to the peak time, Hb A (about 150s), Hb F (about 182s), and Hb A2 (about 243s) were distinguished from other abnormal hemoglobin variants in Zones 7 and 9, and the abnormal hemoglobin can be screened for gene analysis.
2.3.2. Genomic DNA extraction
DNA was extracted according to the operating instructions of the Lab-Aid 820 nucleic acid extraction kit. DNA concentration and purity were detected by the ASP-2680 nucleic acid analyzer, A260/A280 was 1.6-1.9, and the concentration was 20–30 ng/µl.
2.3.3. Common thalassemia genetic testing
Gap-PCR was used to detect 4 deletion types of ɑ-thalassemia in the Chinese population, including –SEA (Southeast Asia), –THAI (Thailand), -α3.7 (rightward), -α4.2 (leftward). RDB was used to detect 3 non-deletion types of ɑ-thalassemia including Hb Constant Spring (HBA2: c.427T > C), Hb Quong Sze (HBA2: c.377T > C), Hb Westmead (HBA2: c .369G > C) and 17 β-thalassemia mutations including codons 41/42 (-TCTT) (HBB: c.126_129delCTTT), IVS-II-654(C > T) (HBB: c.316-197C > T), codon 17 (A > T) (HBB: c.52A > T), −28(A > G) (HBB: c.−76A > G), codon 26 (or Hb E) (G > A) (HBB: c.79G > A), codons 71/72 (+A) (HBB: c. 216_217insA), codon 43 (G > T) (HBB: c.130 G > T), −29 (A > G) (HBB: c.−79A > G), initiation codon (T > G) (HBB: c.2T > G), codons 14/15 (+G) (HBB: c.45_46insG), codons 27/28 (+C) (HBB: c.84_85insC), −32(C > A) (HBB: c.−82C > A), −30(T > C) (HBB: c.−80T > C), IVS-Ⅰ-1(G > T) (HBB: c.92 + 1G > T), IVS-Ⅰ-5(G > C) (HBB: c.92 + 5G > C), CD31(-C) (HBB: c .94delC), CAP + 40-+43.
2.3.4. Sanger sequencing
DNA sequencing was performed by Sanger sequencing. We used NM_000517 as the reference sequence to detect α-globin genes (HBA1, HBA2) and β-globin gene (HBB) for samples with hemoglobin variant detected (). We used 50 ng of genomic DNA and 20 pmol of forward (F) and reverse (R) primers to amplify the PCR apparatus (C1000 Thermal Cycler; Bio-Rad Laboratories) and then sequenced on the ABI PRISM® 3130 automated sequencer (Applied Biosystems). DNA sequencing results were compared with the UCSC database (https://genome.ucsc.edu/) and HBVar database (http://globin.bx.psu.edu/cgi-bin/hbvar/counter) by using Seqman software.
Table 1. Primer sequences of thalassemia gene by Sanger sequencing.
2.3.5. The copy number variation of HBA and HBB genes by MLPA
The SALSA P140-C1 HBA probe of MRC-Holland Company was used to detect the α-thalassemia gene, and the SALSA P102-D1 HBB probe was used to detect the β-thalassemia gene. We used ABI 3500Dx Genetic Analyzer to analyze the amplification product. Cofalyser special software (MRC Holland) was used to analyze the sample data. The normal control ratio was 1.0 ± 0.3. If the ratio is less than 0.7, it means gene deletion, and if it is greater than 1.3, it means gene reiteration.
2.3.6. SMRT and data analysis
Genomic DNA was amplified by PCR with primers that covered the majority of known single nucleotide variations (SNVs), structural variations(SV), insertions and deletions (indels) in the HBA1, HBA2, HBB, HBG and HS40 genes. Using a one-step end-repair and ligation reaction ligated to a barcoded adapter to achiev Prelibrary construction of PCR products. These samples are pooled in equal quantities and converted into the SMRT bell-shaped library by the Sequel Binding and Internal Ctrl Kit 3.0 (Pacific Biosciences). Next, we sequenced the SMRT bell-shaped library by using the CCS mode of the Sequel II platform (Pacific Biosciences), then we converted it to CCS reads, and de-barcoded by lima in the Pbbioconda package (Pacific Biosciences). After the alignment of the processed reads to genome build hg38 by pbmn2 (Pacific Biosciences), we can identify structural variations according to the HbVar, Ithanet and LOVD databases. We use pb amplicon clustering and WhatsHap (version 0.18) software to analyze Haplotype, and use FreeBayes1.3.4 to identify the SNVs and indels.
3. Results
3.1. Hemoglobin electrophoresis analysis
In total, 93 cases of Hb E(in Z4) were detected by the Capillarys2 capillary electrophoresis system, and the diagnosis was confirmed by RDB. There were 81 cases of other rare abnormal hemoglobin in the Hb Zone, we detected 0.73% (174/23709) prevalence of hemoglobinopathy. Rare abnormal hemoglobin in the Hb Zone accounted for 0.34% of the total detected amount (81/23709). They were mainly distributed in the Hb Zone 1(Z1) (2/81), Z3 (1/81), Z5 (2/81), Z7 (12/81), Z6 (5/81), Z8 (1/81), Z11 (45/81), Z12 (8/81) and five cases of hemoglobin variants were detected in the non-single region (). By RDB and Sange sequencing technique, the detection rate of rare hemoglobin variants in this study was 87.65% (71/81). The sensitivity of the capillary electrophoresis apparatus for the detection of Hb NewYork (in Z11), Hb Q-Thailand (in Z7) and Hb Handsworth (in Z5) was 100%, and the sensitivity for the detection of rare hemoglobin variants in Z12 was 62.5% (5/8). It can be seen that Capillarys2 capillary electrophoresis apparatus is highly sensitive to detect various hemoglobin variants, but it is difficult to diagnose the genotype of hemoglobin variants according to the peak region in CE, so it is necessary to identify the specific genotype by combining the gene detection technology.
Table 2. Type and composition ratio of rare hemoglobin variants in the Hb Zone.
3.2. Rare hemoglobin variant detection and clinical phenotype
The molecular detection results and regional distribution of 81 Hb Zone samples with rare hemoglobin variants are shown in . The genotype of 71 cases of hemoglobin variants has been identified, but there are still 10 cases whose genotype could not be identified by the existing detection methods, the detection rate of rare hemoglobin variants by CE was 87.65% (71/81). Hb Cibeles ((A,B)), Hb J-Wenchang-Wuming ((A,B)) and Hb Ube-2 ((A,B)) were detected in Z12, and we found that more types of hemoglobin variants were detected in Z12 than in other regions. The peak time of Hb Cibeles, Hb J-Wenchang-Wuming and Hb Ube-2 was about 102s, 80s and 93s, respectively. The electrophoretic mobility of them was similar, but not completely consistent, indicating that the types of hemoglobin variants detected by capillary electrophoresis in the same region may be different, and it can be roughly distinguished by the peak time. The clinical phenotype and abnormal hemoglobin content of 71 rare hemoglobin variants are shown in , this allows us to study the effect of hemoglobin variants on clinical phenotypes, among which 25 cases of the Hb Zone hemoglobin variants combined with α-thalassemia, 5 cases combined with β-thalassemia, and 1 case combined with α-thalassemia and β-thalassemia ( and ). We found that the abnormal hemoglobin content was correlated with the condition of complex thalassemia.
Figure 1. (A): The hemoglobin variant Hb Cibeles identified by Sanger sequencing. The arrow shows the mutation of the HBA2 gene (B): Hb Cibeles eluted in Z12 on the Capillarys 2 CE analyzer. The arrow shows the abnormal peak.
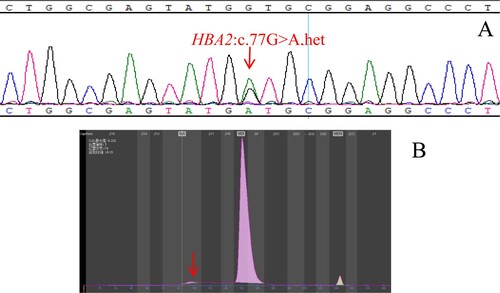
Figure 2. (A): The hemoglobin variant Hb J-Wenchang-Wuming identified by Sanger sequencing. The arrow shows the mutation of the HBA1 gene. (B): Hb J-Wenchang-Wuming eluted in Z12 on the Capillarys 2 CE analyzer. The arrow shows the abnormal peak.
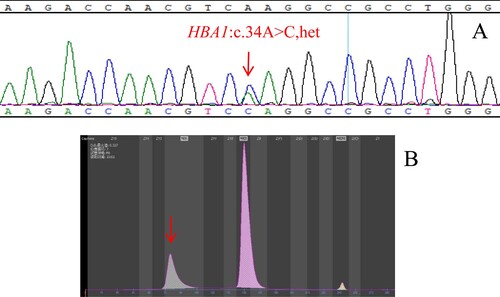
Figure 3. (A): The hemoglobin variants Hb Ube-2 identified by Sanger sequencing. The arrow shows the mutation of the HBA1 gene (B): Hb Ube-2 eluted in Z12 on the Capillarys 2 CE analyzer. The arrow shows the abnormal peak.
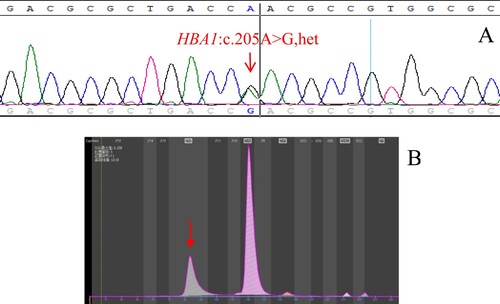
Table 3. The distribution of rare hemoglobin variants.
Table 4. The hematologic phenotype of rare hemoglobin variation in the Hb Zone.
Table 5. Hemoglobin variation in the Hb Zone with thalassemia.
3.3. Rare hemoglobin variant analysis by capillary electrophoresis and molecular diagnostic
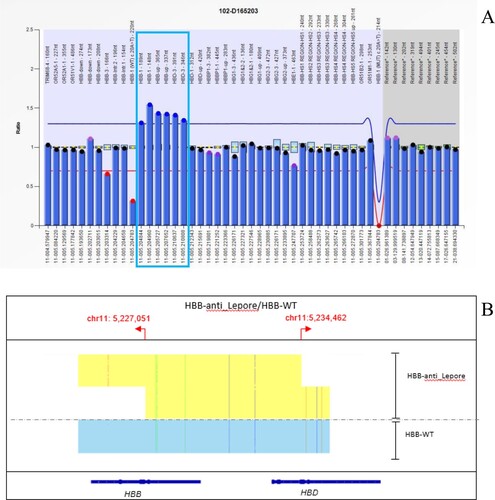
Sanger sequencing and MLPA detection were performed on 81 samples with hemoglobin variants in the Hb Zone (), and rare hemoglobin variant Hb Cibeles was detected ((A,B)), which was the first report in Asia; we found Hb J -Broussais, Hb G-Honolulu, and Hb J-Wenchang-Wuming, which were all first reported in Guangxi. We also found Hb Ube-2, Hb G-Siriraj, Handsworth, Hb Q-Thailand and Hb NewYork. 1 case of 7412 bp duplicate (hg38 CHR11:5,227,051-5,234,462) was identified by SMRT and named Hb Anti-Lepore Liuzhou ((A,B)).
Figure 4. (A): The MLPA(P102) technique detects copy number variation. The proband had a duplicated copy, shown in the blue box region of the β-globin gene cluster. (B): SMRT results of the proband. The arrow shows a 7,412 bp duplicate (hg38 chr11:5,227,051-5,234,462) of the β-globin gene cluster.
3.4. Phenotype comparison of rare hemoglobin variants Hb Cibeles
Hematological phenotypes of Hb Cibeles detected in this study were compared with those in the previous literature (). There were significant differences in MCV, MCH and clinical manifestations between the two cases. Case 1 has micro-erythroemia and shows mild thalassemia, while the hematological parameters of Case 2 were critical, and only showed static thalassemia.
Table 6. Hematologic phenotype of Hb Cibeles carriers.
4. Discussion
Hemoglobinopathy is a kind of hereditary blood disease caused by an abnormal structure or insufficient synthesis of the globin chain. Studies have shown that when the structure of the peptide chain changes, unstable hemoglobin variants can be generated, leading to gene mutation transcription or post-transcription metabolic abnormalities, and the expression of the affected globin chain will decreases, resulting in thalassemia manifestations [Citation13]. In this study, 174 cases of group J, K, Q, G and E hemoglobin variants were found by hemoglobin electrophoresis in Guizhong, Southern China, and the prevalence of hemoglobinopathy was 0.73% (174/23709), higher than that of Meizhou, Guangdong Province (0.477%) [Citation14], Shaokwan, Guangdong Province (0.46%) [Citation15] and Fujian Province (0.26%) [Citation16]. We identified 71 cases of mutations in the Z zone, including 24 cases of α-globin chain hemoglobin variants (Hb Cibeles, Hb J-Broussais, Hb G-Honolulu, Hb J-Wenchang-Wuming, Hb Ube-2, Hb Handsworth, Hb Q-Thailand), and 47 cases of β-globin chain hemoglobin variants (Hb G-Siriraj, Hb NewYork, Hb Anti-Lepore Liuzhou).
We found that the mutation types of different regions in hemoglobin electrophoresis detection are different, and the mutation types of the same region may also be different (). A rare hemoglobin variant Hb Cibeles was detected in Z12 ((A,B)). The molecular structure of Hb Cibeles is extremely unstable [Citation17]. Hb Cibeles detected in our study are reported for the first time in Asia, and only an adopted Ethiopian 4-year-old male carrier reported in 2014 by Felix de la Fuente-Gonzalo [Citation17]. They suggest that the tertiary structure of hemoglobin was affected by unstable Hb Cibeles, leading to the manifestations of nondeletion α-thalassemia. Comparing the hematological phenotypes of two cases (), we found that although they were both Hb Cibeles carriers, the Ethiopian male (Case 1) showed microcytosis and hipocromy [Citation17]. While the male carrier in this study (Case 2) showed static α-thalassemia, with hematological indicators in critical value. Hemoglobin variant content (Hb Z) of about 0.9% was detected, and the manifestations are not as obvious as in Case 1. It may be related to individual differences and low Hemoglobin variant content, but according to the available data, intermediate or severe thalassemia may appear when Hb Cibeles is co-inherited with α-thalassemia. In this study, Hb J-Broussais co-inherited with static α-thalassemia -α3.7/αα was detected, and its Hb Z was 29.1%. In the case of Hb J-Broussais co-inherited with β+ thalassemia [Citation18], Hb Z was 19.4%, the comparison shows that there is a change in Hb Z. Hb J-Wenchang-Wuming, Hb J-Broussais, Hb Ube-2 were also detected in Z12. They cause a few hematological changes, which are consistent with the reported in previous literature [Citation19,Citation20]. Hemoglobin variants above were isolated in Z12. Previous studies also found that a variety of hemoglobin variants were detected in this zone [Citation21,Citation22], indicating that there may be rare hemoglobin variants when Z12 has an abnormal peak, which requires further molecular diagnosis. The hemoglobin variants detected in Z6 were Hb G-Honolulu and Hb G-Siriraj. Hb G-Honolulu was a stable hemoglobin variant. Although this variation affected the physical properties of hemoglobin, it did not change its function [Citation23]. Hb G-Siriraj is a relatively stable hemoglobin, which is the change of external amino acid [Citation24]. Literature shows that Hb G-Siriraj carriers usually have no manifestations [Citation25,Citation26]. The sample in this study(Hb 158 g/L, MCV 70.7 fl, MCH 22.4 pg, Hb A2 4.4%, Hb Z 11.1%) showed mild β-thalassemia, which was inconsistent with previous reports. Zeng [Citation24] first reported a homozygous case of Hb G-Siriraj with microcytic hypochromic anemia. Sanger sequencing detected that the Hb G-Siriraj in this case was heterozygous, and no other HBB gene mutations were detected, so the possibility that homozygous can be ruled out, we need to collect pedigree to further exploration. It can be seen that although most Hb G-Siriraj carriers were normal, it is still necessary to be alert to the possibility of moderate and severe thalassemia in a fetus which is homozygous or when it is co-inherited with β-thalassemia.
Hemoglobin variants co-inherited with α-thalassemia and/or β-thalassemia are more common in Southern China, and it is Hb Z will also change with thalassemia ( and ). Hb Q-Thailand was detected in the Z7 region. In this study, the Hb Z of Hb Q-Thailand carriers was about 17.80%; When Hb Q-Thailand with -α4.2/αα, the Hb Z was 28.55% and its hematological phenotype was critical, with static thalassemia phenotype. When Hb Q-Thailand with -α4.2/HBA2: c.369G > C and HBB: c.−76A > G/WT, the Hb Z was 34.10%, showing mild thalassemia. 45 cases of Hb New York were detected in Z11. Hb Z of Hb New York carriers was about 44.5%, and their hematological phenotypes are variable. Some cases may have microcytic hypochromic anemia, which is due to the slight instability of Hb New York, which may have a slight impact on the physiological state of red blood cells [Citation27]. Hb New York co-inherited with α-thalassemia had a Hb Z of 36-40%, and the abnormalities were consistent with a degree of thalassemia. When Hb New York co-inherited with mild β-thalassemia, Hb Z was 92.05%, and Hb A was not detected, indicating that there was no normal β-chain generated at this time. Since Hb New York still had a chemical function, the body only showed mild β-thalassemia although Hb Z was extremely elevated. Analysis of the data above can speculate that the Hb Z of carriers who co-inherited the same type of thalassemia is usually increased, which may be due to a further decrease in normal globin chain production, leading to the increase in abnormal hemoglobin. However, Hb Z decreased when co-inherited with a different type of thalassemia, and the more severe the thalassemia, the greater the change in Hb Z, which may be due to the proportion change of α- and β-globin chain.
One novel mutation was identified, which was named Hb Anti-Lepore Liuzhou ((A,B)). We identified the location of structural variation by SMRT, HBB-anti-Lepore 7,412 bp duplicate (hg38 Chr11:5,227,051-5,234,462). It is the product of the unequal exchange of gene fusion between the β-chain and δ-chain. In general, the synthesis level of the β-δ fusion gene is much lower than that of a normal β-globin chain but much higher than that of a normal δ-globin chain [Citation28]. The results of routine thalassemia gene detection of the samples in this study were –SEA/αα, RBC 6.44*10^12/L, Hb 137 g/L, MCV 65.9fl, MCH 21.2pg, HbA2 16.5%, an abnormal peak appears in Z3. The peak of hemoglobin variant coincides with HbA2. So far, previously reported Anti-Lepore variants include Hb Parchman, Hb Miyada, Hb P-India, Hb P-Congo, Hb Lincoln Park, Hb P-Nilotic and Hb anti-Lepore Hong Kong [Citation29,Citation30], most of them have normal hematological phenotypes, but only Hb Anti-Lepore Hong Kong (NG_000007.3:g.63160_70572dup) has elevated HbA2 content caused by a high level of β- δ fusion gene expression[Citation28,Citation31]. However, it is not the same breakpoint as Hb Anti-Lepore Liuzhou. A study of So C C [Citation31] showed that Hb Anti-Lepore Hong Kong’s globin gene was closely related to the 5′ locus control region (LCR) and gene expression degree. Hb anti-Lepore Hong Kong co-inherited with β0-thalassemia may lead to intermediate thalassemia. Considering that Hb Anti-Lepore Liuzhou may be similar to Hb anti-Lepore Hong Kong in gene expression, we should pay attention to prenatal diagnosis.
There are some limitations in this study, some of the newly discovered rare hemoglobin variants could not be analyzed by pedigree, and more cases need to be collected for the follow-up study.
In this study, 15 regions divided by hemoglobin electrophoresis were classified. 1 new hemoglobin variant, Hb Anti-Lepore Liuzhou, and 9 rare hemoglobin variants were discovered and found in Z1-Z12. It is worth noting that although most of the hemoglobin variants have normal hematological phenotypes, we found that Hb Cibeles and Hb G-Siriraj have different hematological phenotypes from previous studies, when they co-inherited with thalassemia may lead to moderate or severe anemia. More data should be collected for further research. We found that some hemoglobin variants may affect other indicators, and we need to screen them in daily detection work. This study deepened our understanding of co-inheritance between hemoglobin variants and thalassemia and provided important data support for prenatal counseling and prenatal diagnosis by analyzing hemoglobin variants.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Hu TT, Guo H, Qin DQ, et al. Molecular diagnosis and hematological characteristics of abnormal hemoglobin J-Bangkok. Chin J Clin Lab Sci. 2020;38(03):171–174.
- Xie JH, Wang GQ, Xiao QZ, et al. Screening and molecular diagnosis for abnormal hemoglobin. Lab Med. 2016;31(01):22–25.
- Taher AT, Weatherall DJ, Cappellini MD. Thalassaemia. Lancet. 2018;391(10116):155–167.
- Ren TF. Screening analysis of abnormal hemoglobin disease in people of childbearing age in Xinyi district, guangdong province. Chin J Mod Drug Appl. 2021;15(06):240–242.
- Pang YM, Wang XC, Pan HF, et al. A clinical study on genotype, hematological phenotype and iron overload of 1110 patients with hemoglobin H disease in Guangxi. Chin Youjiang Med J. 2019;47(05):336–342.
- Keikhaei B, Rahim F, Zandian KH, et al. Comparison of different indices for better differential diagnosis of deficiency anemia from thalassemia trait. Blood. 2007;4(1):95.
- He Y, Zhang YH, Wu RX, et al. Value of the combined tests of mean corpuscular volume, red cell osmotic fragility test and capillary hemoglobin electrophoresis in antenatal screening and diagnosis of thalassemia. Int J Lab Med. 2013;34(19):2521–2523 + 2525.
- Deng YY, Li Y. Analysis the diagnostic value of Gap PCR and PCR-fluorescence probe in thalassemia. Gansu Med J. 2020;39(7):622–623.
- Xiao QZ, Wang G, Li LX, et al. Verification application of PCR techniques with different principles in prenatal diagnosis of thalassemia. J Pract Med. 2017;33(6):994–996.
- Jiao BQ, Li W, Liu F. Multiplex ligation-dependent probe amplification gene detection for diagnosis of 3 patients with Prader-Willi syndrome. Chin J Appl Clin Pediatrics. 2016;31(20):1580–1581.
- Lv LN, Yang CH. Diagnosis and treatment status of thalassemia and molecular biology research. J Med Inf. 2022;35(11):33–36.
- Tomonari M, Shun M, Masami Y. Mutation assay using single-molecule real-time (SMRT(TM)) sequencing technology. Genes Environ Official J Jpn Environ Mutagen Soc. 2015;37(1):15.
- Xu XM. Guidelines of thalassenmia prevention and control programme. Beijing: People’s Military Medical Press; 2011.
- Lin M, Wen YF, Wu JR, et al. Hemoglobinopathy: molecular epidemiological characteristics and health effects on Hakka people in the Meizhou region, southern China. Plos One. 2013;8(2):e55024.
- Yang H, Lin M, Lin F, et al. Molecular epidemiological investigation of abnormal hemoglobin in Shaokwan region, Southern China. Hematology. 2022-12-01;27(1):150–156.
- Huang HL, Xu LP, Chen MH, et al. Molecular characterization of thalassemia and hemoglobinopathy in southeastern China. Sci Rep. 2019;9(1):3493.
- de la Fuente-Gonzalo F, Nieto JM, Vinuesa L, et al. Hb cibeles [α2 CD25(B6) (Gly → Asp)]: a novel alpha chain variant causing alpha-thalassemia. Int J Hematol. 2014;100(6):599–601.
- Durou MR, Ribault A, Dufour MJ, et al. Fortuitous diagnosis of the association of hemoglobin J-Broussais with beta + thalassemia. Ann Biol Clin. 1996;54(8-9):321–323.
- Zhai YS, Tang HS, Li DZ. Hb J-Wenchang-Wuming [α11(A9)Lys→Gln (AAG > CAG) (α2 or α1)] compromises neonatal screening for α-thalassemia with the sebia Capillarys2 electrophoresis system. Hemoglobin. 2012;36(4):395–398.
- Chen QQ, Xian JJ, Wang YC, et al. Molecular diagnosis and pedigree analysis of abnormal hemoglobin in hemoglobin UBE-2 combined with CD37 heterozygous. J Pract Med. 2022;38(4):5.
- Panyasai S, Kongthai K, Phasit A. Association of Hb Shenyang [α26 (B7) Ala→Glu, GG > GG,: c.80C > A (or)] with several types of α-thalassemia in Thailand. Hemoglobin. 2020;44(5):354–360.
- Recasens V, Montaés N, Rodríguez-Vigil C, et al. Why is the novel Hb miguel servet visualised by CE-HPLC newborn and not by the CE-HPLC β-thalassaemia programme?. J Clin Pathol. 2021;74(3):198–201.
- Paleari R, Caruso D, Giavarini F, et al. The first case of Hb G-honolulu [α30(B11)Glu→Gln (GAG > CAG); HBA2:c.91G > A] observed in association with Hb S [β6(A3)Glu→Val, GAG > GTG] in a healthy Italian child. Hemoglobin. 2012;36(1):73–79.
- Zeng YT, Huang SZ. Studies on Hb siriraj homozygote. J Genet Genomics. 1979;02:3-6 + 83–84.
- Li YM, Jin X, Zhang YY, et al. Molecular characteristics and hematological phenotype of hemoglobin G in Shenzhen. Chin J Clin Lab Sci. 2020;38(03):180–183.
- Wang YF, Wu BY, Xia WQ, et al. Hematologic phenotype and genotype analysis of patients with hemoglobin variants. J Exp Hematol. 2021;29(04):1280–1288.
- Cheng ZR, Zhu XN, Zeng D, et al. Hb New York, preliminary results concerning the hematologic characteristics and the effects on thalassemia. Mol Biol Rep. 2022;49(7):6199–6205.
- Elisabeth S, Kamran M, Fabrizio D, et al. Comparison of two known chromosomal rearrangements in the δβ-globin complex with identical DNA breakpoints but causing different Hb A(2) levels. Hemoglobin. 2012;36(2):177–182.
- Efremov GD. Hemoglobins lepore and anti-Lepore. Hemoglobin. 2009;2(3):197–233.
- Driscoll MC, Ohta Y, Nakamura F, et al. Hemoglobin miyada: DNA analysis of the anti-Lepore βδ fusion gene. Am J Hematol. 1984;17(4):355–362.
- So CC, Chan A, Tsang S, et al. A novel beta-delta globin gene fusion leads to over-expression of delta globin chain and a mild thalassemia intermedia phenotype when Co-inherited with β0-thalassemia. Blood. 2006;108(11):1587–1587.