
ABSTRACT
Objectives:
This study aimed to evaluate the incidence and prognostic significance of common cytogenetic and molecular abnormalities in patients with TP53-mutated and non-TP53-mutated acute myeloid leukemia (AML).
Methods:
We retrospectively analyzed the clinical data of 326 patients with newly diagnosed AML hospitalized in our institution between October 2015 and June 2021. Classification variables were reported as percentages and compared by χ2 tests. Survival rates were evaluated by the Kaplan-Meier method.
Results:
The incidence of TP53 mutations in AML patients in this clinic was 9.8%, of whom 87.5% patients were over 50 years old. The common concurrent mutations of TP53 were DNMT3A, IDH2, NRAS and TET2. Patients with a TP53 variant allele frequency (VAF) ≤ 40% had better overall survival (OS) than patients with a VAF >40%. Compared with non-TP53-mutated patients, significantly more TP53-mutated patients were gene-fusion negative, +mar, – 7/del (7q), – 5/del (5q), – 17/17p-, – 12/12p-, incomplete (inc) karyotype, or complex karyotype (CK), and had FLT3-ITD or IDH2 mutations, as well as a lower complete remission (CR) rate (31.3%) and higher recurrence rate (80.0%). The 2-year OS rates of TP53-mutated and non-TP53-mutated patients were 18.8% and 47.3%, respectively (P < 0.001). Univariate analysis showed that non-TP53-mutated patients with MLL family gene fusion, +mar or – 17/17p – karyotype, and FLT3-ITD mutations had a poor prognosis, while t(8; 21) karyotype was associated with a better prognosis. TP53-mutated patients with – 7/del (7q) or – 5/del (5q) karyotype had a poor prognosis.
Conclusions:
The cytogenetic and molecular landscapes differed between TP53-mutated and non-TP53-mutated patients, and some abnormalities had different values between them.
Introduction
Acute myeloid leukemia (AML) is a common hematological malignant tumor with a highly heterogeneous prognosis, which may be driven by the expansion of previously established genetic abnormalities or the emergence of new abnormalities. AML has been classified according to the World Health Organization (WHO) classification, and genetic abnormalities have been incorporated into the diagnostic algorithms for AML. Recurrent genetic abnormalities include balanced translocations (t)/inversions (inv) and gene mutations, which are present in 20% – 30% of patients with AML. AML with balanced translocations may include t(8; 21)(q22; q22.1), t(3; 3)(q21.3; q26.2), and t(1; 22)(p13.3; q13.1)[Citation1]. Some rare abnormalities, such as pediatric AML-M5 with MLL-SEPT6 fusion, have favorable outcomes, whereas variant RARA rearrangement, t(12; 19)(q13; q13), t(4; 8)(q32; q13), and t(4; 11)(q21; p15) are usually associated with poor prognosis.[Citation2–5].
Cytogenetic and molecular abnormalities may affect treatment efficacy and prognosis, and their occurrence and prognostic significance show diverse characteristics in different populations. Research into the prognosis of AML patients remains a hot topic. Improved understanding of the molecular characteristics of AML and the recent development of next-generation gene sequencing (NGS) mean that molecular detection, based on gene expression, has improved the prognostic evaluation and treatment strategies of AML. Most molecular abnormalities, such as TP53 mutations, affect the prognosis of AML but lack specific treatments, and their occurrence are mostly related to age [Citation6–8]. TP53 mutations are one of the most common alterations in cancer. TP53 plays important regulatory roles in apoptosis and cell cycle integrity, and its inactivation may lead to uncontrolled cell proliferation and promote cancer development [Citation9]. Approximately 5% – 10% of newly diagnosed cases of AML have mutations in the TP53 gene, which tends to be associated with a poor prognosis [Citation10–16].
In this study, we investigated the clinical characteristics, cytogenetic and molecular landscapes, probability and diversity of co-occurring mutations, and prognostic significance of these abnormalities in AML patients with and without TP53 mutations in a real-world setting.
Materials and methods
Patients and diagnosis
We retrospectively analyzed 326 patients with newly diagnosed AML and available cytogenetic and molecular data, who received treatment at The Affiliated Huaian No.1 People's Hospital of Nanjing Medical University between October 2015 and June 2021. AML was diagnosed according to the 2016 revision of the WHO classification of myeloid neoplasms and acute leukemia [Citation17]. Patients with acute promyelocytic leukemia and patients lacking baseline mutation data were excluded. The clinical characteristics of the patients were collected, including age, sex, white blood cell count (WBC), hemoglobin (Hb), platelet count (PLT), cytogenetic and molecular abnormalities, initial and subsequent treatment strategies, and overall survival (OS). Bone marrow examination was performed before each consolidation chemotherapy or when the disease condition changed. All the patients provided written informed consent. The study was approved by the relevant ethics committee and was performed in accordance with the Declaration of Helsinki.
Cytogenetic and molecular examination
Morphological, immunological, cytogenetic, and molecular biological examinations were performed at the initial diagnosis of AML. Real-time quantitative polymerase chain reaction (RT-qPCR, ELISA Kit) was used to screen 45 common fusion genes, including CBFB-MYH11, BCR-ABL1, RUNX1-RUNX1T1, and other gene fusions involving ETV6 (ETS variant 6), MLL (mixed lineage leukemia), and HOX family members. Cytogenetic examination was performed with conventional karyotyping (R-banding). According to the International System for Human Cytogenomic Nomenclature (ISCN 2020), clonal structure and numerical chromosome abnormalities were reported. Molecular abnormalities were detected by NGS using a 15-gene panel at the time of initial diagnosis. The 15-gene panel included FLT3 (fms-like tyrosine kinase 3), NRAS (neuroblastoma RAS viral oncogene homolog), C-KIT (KIT proto-oncogene receptor tyrosine kinase), WT1 (Wilms tumor 1), TET2 (tet methylcytosine dioxygenase 2), IDH1 (isocitrate dehydrogenase 1), IDH2 (isocitrate dehydrogenase 2), DNMT3A (DNA methyltransferase 3a), ASXL1 (additional sex combs like 1), NPM1 (nucleophosmin 1), CEBPA (CCAAT/enhancer binding protein a), RUNX1 (runt-related transcription factor 1), PHF6 (PHD finger protein 6), SF3B1 (splicing factor 3b subunit 1), and TP53. DNA was sequenced using a Nextseq550 system (NextSeq Sequencer, America) with probe capture and database building, with an average sequencing depth ≥1000×.
Treatment and efficacy evaluation criteria
A total of 326 patients with AML received frontline treatment, including 204 patients who received induction chemotherapy with intravenous (i.v.) cytosine arabinoside (Ara-C, 100 mg/m2/day) for 7 consecutive days, together with idarubicin (I, 10–12 mg/m2/day, i.v.) or daunorubicin (60 mg/m2/day, i.v.), or homoharringtonine (H, 2.5 mg/m2/day, i.v.) for 3 consecutive days (‘3 + 7’ regimen), and 122 unfit patients who received low-intensity treatment with azacitidine (75 mg/m2) subcutaneously for 7 consecutive days combined with venetoclax (100 mg day 1, 200 mg day 2, 400 mg days 3–28, orally), IAG (I 10 mg days 1–3, i.v.; Ara-C 10 mg/m2, once every 12 h, days 1–7, i.v.; granulocyte colony-stimulating factor 300 µg/day until WBC ≥20 × 10^9/L, i.v.) or HAG (H 1 mg/m2, days 1–7, i.v.; Ara-C 10 mg/m2, once every 12 h, days 1–7, i.v.; granulocyte colony-stimulating factor 300 µg/day until WBC ≥20 × 10^9/L, i.v.). Consolidation treatment included cytarabine (1–3 g/m2/day, days 1–3); original induction scheme, autotransplantation, and allogeneic hematopoietic stem cell transplantation.
Complete remission (CR) and morphologic leukemia-free state were defined according to the European Leukemia Network (ELN 2017) criteria, and grouping was defined by the ELN 2017 criteria for combined gene mutations and karyotype abnormalities [Citation18]. CR referred to bone marrow blasts <5%, neutrophil count ≥1 × 10^9/L, PLT ≥100 × 10^9/L, and no evidence of extramedullary leukemia. OS was defined as the time from diagnosis to death from any cause or censored at last follow-up.
Statistical analysis
Data were analyzed using SPSS version 17.0 (SPSS, Inc., Chicago, IL, USA). Continuous variables were expressed as median and range and compared using t-tests, and categorical variables were reported as percentages and compared using χ2 tests. OS rates, determined from diagnosis to death, were estimated by the Kaplan – Meier method and curves were compared using log-rank analysis. Risk factors was analyzed by Cox regression. A p value <0.05 was considered to indicate a significant difference.
Results
Baseline characteristics of patients with and without TP53 mutations
A total of 326 out of 396 AML patients were available for analysis, including 294 patients without TP53 mutations and 32 with TP53 mutations. The average age of the 326 patients was 57 years (range, 14–88 years) and the median follow-up time was 10 months (range, 1–67 months). The median survival times were 6 months (range, 1–47 months) for the 32 TP53-mutated patients and 11.4 months (range, 1–67 months) for the 294 non-TP53-mutated patients. The basic clinical characteristics of the two groups of patients are listed in . M2 and M5 were the most common subtypes, and there were no significant differences in sex, WBC, Hb, and PLT between the TP53-mutated and non-mutated patients. The proportion of peripheral blood primordial cells was higher in non-mutated patients. TP53 mutations were more common in patients aged ≥50 years(87.5%) (P < 0.001). Patients with TP53 mutations were all high-risk according to the 2017 ELN criteria. These patients were also more likely to be fusion-gene negative; to have + mar (marker chromosome), – 7/del (7q), – 5/del (5q), – 17/17p-, – 12/12p-, and inc karyotypes (one or more incomplete karyotypes), CK (complex karyotype, three or more abnormalities), and FLT3-ITD mutations; and to have a lower CR rate (31.3%) and higher recurrence rate (80.0%). Patients without TP53 mutations were more likely to be low – or medium-risk according to the 2017 ELN stratification, to have higher probabilities of t(8; 21) karyotype and IDH2 mutations, to receive standard chemotherapy, and to have a higher CR rate (58.8%) and lower recurrence rate (56.6%).
Table 1. Baseline characteristics of AML patients with and without TP53 mutations.
OS of patients with and without TP53 mutations and the other prognostic factors
The 1-year OS rates of TP53-mutated and non-mutated AML patients were 28.1% and 58.8%, respectively (P < 0.001), and the equivalent 2-year OS rates were 18.8% and 47.3%, respectively (P < 0.001) (). The influences of common cytogenetic and molecular abnormalities on OS were analyzed by univariate analysis. The results showed that MLL family gene fusion (notably DulMLL), +mar and – 17/17p – karyotypes, and FLT3-ITD mutations were associated with a poor prognosis in non-TP53-mutated patients, while t(8; 21) karyotype was associated with a better prognosis (, A-F). The inc karyotype was associated with a poor prognosis, but the difference was not significant (P = 0.077). – 7/del(7q) and – 5/del(5q) karyotypes, CK, and IDH1 mutations were associated with a poor prognosis in patients with TP53 mutations (A-D). Among all 326 cases, RUNX1-RUNX1T1 gene fusion/t(8; 21) karyotype was associated with better prognosis, while DupMLL gene fusion, +mar, – 7/del (7q), – 5/del (5q), – 17/17p-, and – 12/12p-karyotypes, CK, FLT3-ITD and TP53 mutations were associated with poor prognosis (A-J). In addition, MLL family gene fusion (P = 0.060), CEBPA double mutations (P = 0.090), and PHF6 mutations (P = 0.075) were also found to have a prognostic impact, but the findings were not significant.
Figure 1. The 2-year OS of patients with and without TP53 mutations.
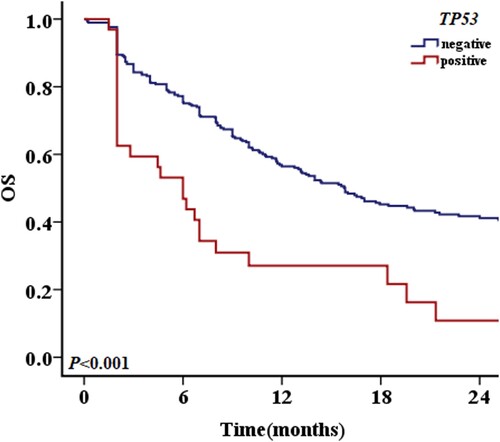
Figure 2. The 2-year OS with different prognostic factors in patients without TP53 mutations. A: OS in patients with and without MLL family gene fusion. B: OS in patients with and without + mar karyotypes. C: OS in patients with and without – 17/17p karyotype. D: OS in patients with and without FLT3-ITD mutations. E: OS in patients with and without DupMLL gene fusion. F: OS in patients with and without t (8; 21) karyotype.
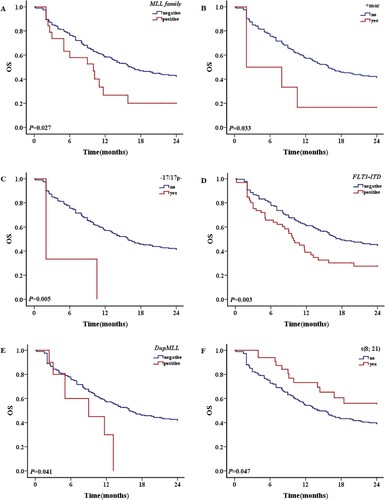
Figure 3. The 2-year OS with different prognostic factors in patients with TP53 mutations. A: OS of patients with and without – 7/del (7q) karyotype. B: OS of patients with and without – 5/del (5q) karyotype. C: OS of patients with and without CK. D: OS of patients with and without IDH1 mutations.
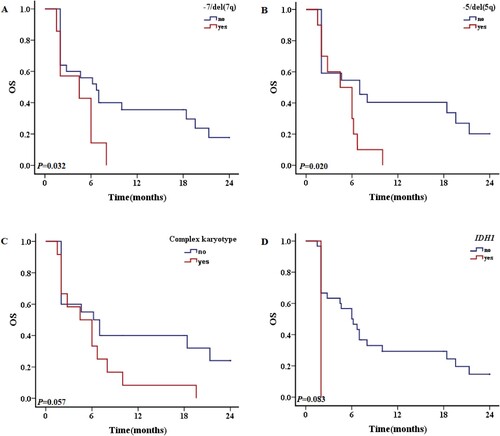
Figure 4. The 2-year OS with different prognostic factors for all patient. A: OS of patients with and without RUNX1-RUNX1T1 gene fusion. B: OS of patients with and without + mar karyotypes. C: OS of patients with and without – 7/del (7q) karyotype. D: OS of patients with and without – 5/del (5q) karyotype. E: OS of patients with and without – 17/17p karyotype. F: OS of patients with and without – 12/12p karyotype. G: OS of patients with and without inc karyotypes. H: OS of patients with and without CK. I: OS of patients with and without FLT3/ITD mutations. J: OS of patients with and without DupMLL gene fusion.
Table 2. Univariate analysis of 2-year OS in patients with and without TP53 mutations.
Multivariate Cox regression analysis showed that age (P < 0.001), treatment strategy (P < 0.001), CR (P < 0.001), relapse (P = 0.007), secondary AML (P = 0.008), M6 subtype (P = 0.011), MLL family gene fusion (P = 0.027), +mar karyotype (P = 0.035), – 17/17p karyotypes (P = 0.006) and FLT3-ITD mutations (P = 0.003) were significantly associated with OS in non-TP53-mutated patients. In addition, t(8; 21) karyotype (P = 0.052) was a relatively good prognostic factor and inc karyotype (P = 0.079) was a relatively poor prognostic factor in non-TP53-mutated patients; however, the findings were not significant. Age (P = 0.027), treatment strategy (P = 0.031), CR (P = 0.014), and – 5/del(5q) karyotype (P = 0.037) were significantly associated with OS in TP53-mutated patients. No other common cytogenetic or molecular abnormality, other than the – 5/del(5q) karyotype, had any significant impact on prognosis. CK (P = 0.083) and – 7/del(7q) (P = 0.055) karyotype were relatively poor prognostic factors in TP53-mutated patients; however, the findings were not significant.
Given that age was an important factor affecting the therapy and prognosis of patients with AML, we focused on the survival of patients in different age groups (A). Patients aged <30 years, 30–39 years, and 40–49 years had significantly better OS than patients aged 50–59 years, 60–69 years, 70–79 years, and ≥80 years. Patients aged ≥50 years were a high-risk population with similar OS rates. Taking the age 50 years as the cut-off, 104 patients were <50 years and 222 were ≥50 years old. The 1-year OS rates were 75.0% and 46.8%, respectively (P < 0.001), and the 2-year OS rates were 68.3% and 33.3%, respectively (P < 0.001). OS was significantly better in patients aged <50 years compared with patients ≥50 years(B). Among patients aged ≥50 years, there was a significant difference in OS between TP53-mutated (n = 28) and non-TP53-mutated (n = 194) patients. However, among patients aged <50 years(n = 104), there was no significant difference in survival between patients with (n = 4) and without(n = 100) TP53 mutations(P = 0.924), possibly because of the small number of cases.
Figure 5. Age distribution and 2-year OS of AML patients in different age groups. A: Distribution of patients in different age groups. B: The OS of patients of different ages. C: OS of patients aged ≥50 years and <50 years.
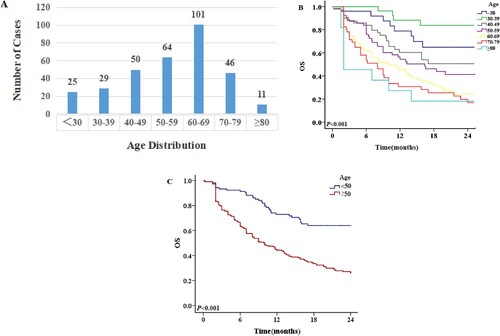
OS of patients receiving a standard or low-intensity regimen
Thirty-two AML patients had TP53 mutations, of whom 21 received low-intensity chemotherapy and 11 received standard ‘3 + 7’ regimen chemotherapy, with CR rates of 23.8% (5/21) and 45.5% (5/11), respectively. The 1-year OS rates of patients receiving low-intensity and standard chemotherapy were 19.0% and 45.5%, respectively (P = 0.241), and the 2-year OS rates were 14.3% and 27.3%, respectively (P = 0.325). Of the 32 patients, 10 achieved CR and 22 did not. Their respective 1-year OS rates were 70.0% and 9.1% (P = 0.002), and the 2-year OS rates were 40.0% and 9.1% (P = 0.008), showing that CR was associated with better survival (B).
Figure 6. The 2-year OS of patients with TP53 mutations. A: OS of patients with different treatment regimens. B: OS of patients weather CR or nor.
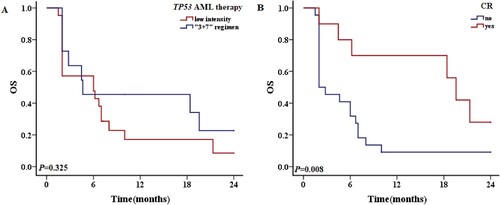
Of the 294 patients without TP53 mutations, 101 received low-intensity chemotherapy and 193 received standard ‘3 + 7’ regimen chemotherapy, with CR rates of 43.6% (44/101) and 72.0% (139/193), respectively. The respective 1-year OS rates of patients receiving low-intensity and standard chemotherapy were 36.6% and 70.5% (P < 0.001), and the 2-year OS rates were 22.8% and 60.1% (P < 0.001) (A). Among 294 non-TP53-mutated patients, 173 achieved CR and 121 did not, and the respective 1-year OS rates were 82.7% and 24.8% (P < 0.001), and the 2-year OS rates were 69.9% and 14.9% (P < 0.001) (B).
Figure 7. The 2-year OS of patients without TP53 mutations. A: OS of patients with different treatment regimens. B: OS of patients weather CR or nor.
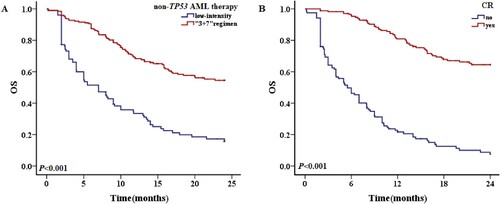
Concurrent mutations in TP53-mutated patients and their effects on survival
Twelve patients (12/32, 37.5%) had TP53 variants alone and 20 patients had other mutations in addition to TP53 mutations. The survival of patients with and without additional mutations were similar (1-year OS rate 30.0% vs. 25.0%, P = 0.472; 2-year OS rate 20.0% vs. 16.7%, P = 0.348) (A). The most common concurrent mutations were DNMT3A (P = 0.003), IDH2 (P = 0.012), NRAS (P = 0.044) and TET2 (P = 0.044). TP53-mutated AML tended to be associated with a lower incidence of FLT3-ITD, CEBPA, WT1 or IDH1 mutations (, ). Patients with a TP53 variant allele frequency (VAF) ≤ 40% had better OS than those with a VAF >40%, with respective 1-year OS rates of 66.7% and 19.2% (P = 0.091), and 2-year OS rates of 66.7% and 7.7% (P = 0.022) (B). The median OS of patients with a TP53 mutation VAF >40% was 5.3 months, compared with 8.1 months in patients with a VAF ≤40%. The frequencies of gene mutations among patients are summarized in . An oncoplot of the characteristics, concurrent mutations, treatments, and outcomes of TP53-mutated AML patients (n = 32) was shown in .
Figure 8. The 2-year OS of patients with TP53 mutations in different situations. A: Patients with and without concurrent mutations in addition to TP53 mutations. B: OS of TP53-mutated patients VAF ≥40% and <40%.
Figure 9. A chart showing the characteristics, concurrent mutations, treatment, and outcomes of TP53-mutated patients. Each column represented one patient, and the presence of aberrations was represented by different colors.
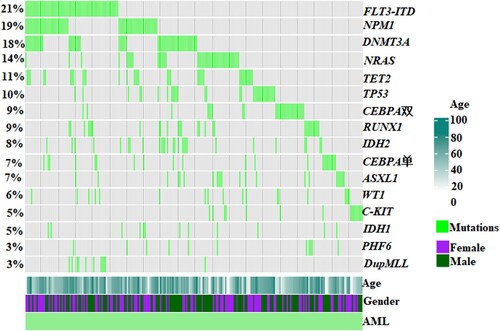
Table 3. Concurrent mutations in TP53-mutated patients.
Discussion
Cytogenetic and molecular abnormalities have received increasing attention in terms of their roles in AML prognosis. TP53 mutations are somewhat rare in AML, but TP53-mutated patients have been identified as an important molecula subgroup with dismal outcomes. The current study showed that the unique clinical biological characteristics of TP53-mutated AML patients were often accompanied by fusion-gene negativity, +mar, – 7/del (7q), – 5/del (5q), – 17/17p-, – 12/12p-, and inc karyotypes, and CK, with a poor treatment response and a higher recurrence rate. However, significantly more non-TP53-mutated patients showed low – and medium-risk in the 2017 ELN stratification, t(8; 21) karyotype, and IDH2 mutations, and more patients received standard chemotherapy, with a higher CR rate and lower recurrence rate. Among the adverse prognostic factors in patients with TP53 mutations, CK occurred in 10% – 15% of adult AML cases and was closely associated with TP53 mutations. TP53 mutations and deficiency were previously shown to allow the development of karyotype aberrations and heterogeneity in myeloid cells by perpetuating chromosome segregation errors [Citation19–21]. Alwash et al. found that TP53 mutations emerged during treatment in 15% of refractory or relapsed AML patients without initial TP53 mutations and in 45% of patients with CK. Patients with CK, chromosome 5 abnormality or IDH2 mutations at initial diagnosis, intensive treatment, and hematopoietic stem cell transplantation were more likely to have TP53 mutations (P < 0.05) [Citation15]. In the current study, patients with TP53 mutations had a high incidence of other karyotypes with a poor prognosis, in addition to CK, which was one of the adverse prognostic factors in these patients. Among TP53-mutated patients in our center, 37.5% had CK, which was a significantly higher percentage than in non-TP53-mutated patients. The present study also showed that TP53 mutations were more common in patients aged ≥50 years, with a lower response rate to chemotherapy, and the efficacy did not depend on the type or intensity of chemotherapy drugs. Even high-dose chemotherapy failed to improve the prognosis of patients with TP53 mutations, which was consistent with other studies [Citation19, Citation22–26]. Notably, even allogeneic hematopoietic stem cell transplantation had limited significance in patients with TP53-mutant AML [Citation13, Citation22]. Patients who achieved complete or partial remission after frontline treatment had better survival than patients without remission, irrespective of transplantation.
In 2020, Short et al. explored the prognostic impact of the TP53 VAF on AML [Citation14]. In multivariate analysis, a TP53-mutated VAF >40% was independently associated with a significantly higher cumulative recurrence rate (P = 0.003) and poorer recurrence-free survival (P = 0.001) and OS (P = 0.003). The median OS of patients with a TP53-mutation VAF >40% was 4.7 months, compared with 7.3 months in patients with a VAF ≤40% (P = 0.006). In our study, patients with a VAF ≤40% had better OS than those with a VAF >40% (P = 0.024), with median OS durations of 8.1 and 5.3 months, respectively, which were consistent with Short et al.'s findings. However, other researchers found different VAF values. Alwash et al. found that a TP53 VAF <20% was associated with an increased likelihood of CR or CR with incomplete hematologic recovery (CRi) compared with a higher VAF (VAF <20%, CR/CRi rate 41%; VAF ≥20%, CR/CRi rate 13%). The median survival times of the two groups were 6.1 and 3.5 months, respectively (P < 0.05), and the prognosis was poor [Citation15].
The concurrent-mutation status and mutual influence of TP53 mutations and other AML mutations remained unclear. TP53 mutations rarely co-existed with FLT3-ITD, CEBPA, or IDH1 mutations, and the most common concurrent mutations were DNMT3A, IDH2, NRAS and TET2, which were consistent with other retrospective studies [Citation15, Citation16, Citation22, Citation27]. However, concurrent mutations were enriched in patients with AML marked by a non-CK. The majority of concurrent mutations are known to contribute to age-related clonal hematopoiesis, which has shown a lack of prognostic significance [Citation20, Citation22, Citation27].
TP53-mutated AML still lacks targeted drugs, and shows inferior responses to high-intensity chemotherapy, hypomethylating agents, venetoclax, CPX-351 (a liposomal form of cytarabine and daunorubicin), and immunotherapy. The response rates of patients with TP53 mutation range from 20% to 42%, with a median OS of 4–9 months [Citation28, Citation29]. Outcomes of allogeneic hematopoietic stem cell transplantation in the TP53-mutated population also tend to be among the poorest, with a 2-year OS of 24.9% and lower relapse-free survival of 23.7% [Citation28, Citation29]. New treatment strategies are needed to overcome the adverse prognosis of TP53 mutations. Klimovich et al. have modeled partial TP53 reactivation by using knock-in mice with inducible expression of the TP53 variant E177R. Owing to the high sensitivity of cancer cells to even small changes in TP53 activity, partial p53 reactivation was sufficient to induce cancer regression [Citation30].
Conclusion
This study found that TP53 mutations were more common in AML patients aged ≥50 years. TP53-mutated patients had specific clinicobiological features and cytogenetic changes associated with dismal outcomes. Newer molecular targeted therapies are urgently needed to enhance the remission rate and outcomes in patients with TP53-mutated AML.
Authors contributions
Hong Liu, Yuye Shi: Contributed to the concept development, study design, and helped write the manuscript. Shandong Tao, Yunjie Li: Collected the clinical information. Chunling Wang, Liang Yu: Contributed to the diagnosis and treatment of patients and approved the final manuscript.
Ethics approval and consent to participate
This study protocol was reviewed and approved by Institutional Review Board (approval number KY-2022-032-01), and was conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from all individual participants included in the study.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Hwang SM. Classification of acute myeloid leukemia. Blood Res. 2020;55(S1):S1–S4.
- Chebly A, Djambas Khayat C, Yammine T, et al. Pediatric M5 acute myeloid leukemia with MLL-SEPT6 fusion and a favorable outcome. Leuk Res Rep. 2021;16:100277.
- Liu G, Liu L, Bartolo DD, et al. Acute promyelocytic leukemia with rare genetic aberrations: a report of three cases. Genes(Basel). 2022;14(1):46.
- Chebly A, Haddad FG, Bassil J, et al. A rare case of acute myeloid leukemia with t(12;19)(q13;q13). Leuk Res Rep. 2020;14:100216.
- Milczarek S, Studniak E, Baumert B, et al. Case of patient with AML with complex karyotype including ultra-rare t(4;8)(q32;q13), t(4;11)(q21;p15) and familial aggregation of myeloid malignancies. Medicina (Kaunas). 2022;58(1):105.
- Creutzig U, Zimmermann M, Reinhardt D, et al. Changes in cytogenetics and molecular genetics in acute myeloid leukemia from childhood to adult age groups. Cancer. 2016;122(24):3821–3830.
- Bolouri H, Farrar JE, Triche T Jr, et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specifific mutational interactions. Nat Med. 2017;24(1):103–112.
- Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classifification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209–2221.
- Kastenhuber ER, Lowe SW. Putting p53 in context. Cell. 2017;170(6):1062–1078.
- Bykov VJN, Eriksson SE, Bianchi J, et al. Targeting mutant p53 for efficient cancer therapy. Nat Rev Cancer. 2018;18(2):89–102.
- Prokocimer M, Molchadsky A, Rotter V. Dysfunctional diversity of p53 proteins in adult acute myeloid leukemia: projections on diagnostic workup and therapy. Blood. 2017;130(6):699–712.
- Kuykendall A, Duployez N, Boissel N, et al. Acute myeloid leukemia: the good, the bad, and the ugly. Am Soc Clin Oncol Educ Book. 2018;38:555–573.
- Niparuck P, Police P, Noikongdee P, et al. TP53 mutation in newly diagnosed acute myeloid leukemia and myelodysplastic syndrome. Diagn Pathol. 2021;16(1):1–8.
- Short NJ, Montalban-Bravo G, Hwang H, et al. Prognostic and therapeutic impacts of mutant TP53 variant allelic frequency in newly diagnosed acute myeloid leukemia. Blood Adv. 2020;4(22):5681–5689.
- Alwash Y, Khoury JD, Tashakori M, et al. Development of TP53 mutations over the course of therapy for acute myeloid leukemia. Am J Hematol. 2021;96(11):1420–1428.
- Li Y, Wan H, Jing Y. Molecular characterization and clinical treatment of acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS) patients with TP53 mutation. Cl Lymph Myelom Leuk. 2021;21(12):841–851.
- Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405.
- Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–447.
- Cazzola A, Schlegel C, Jansen I, et al. TP53 deficiency permits chromosome abnormalities and karyotype heterogeneity in acute myeloid leukemia. Leukemia. 2019;33(11):2619–2627.
- Rücker FG, Schlenk RF, Bullinger L, et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood. 2012;119(9):2114–2121.
- Stengel A, Kern W, Haferlach T, et al. The impact of TP53 mutations and TP53 deletions on survival varies between AML, ALL, MDS and CLL: an analysis of 3307 cases. Leukemia. 2017;31(3):705–711.
- Ciurea SO, Chilkulwar A, Saliba RM, et al. Prognostic factors influencing survival after allogeneic transplantation for AML/MDS patients with TP53 mutations. Blood. 2018;131(26):2989–2992.
- Hou HA, Chou WC, Kuo YY, et al. TP53 mutations in de novo acute myeloid leukemia patients: longitudinal follow-ups show the mutation is stable during disease evolution. Blood Cancer J. 2015;5(7):E331–E331.
- Kadia TM, Jain P, Ravandi F, et al. TP53 mutations in newly diagnosed acute myeloid leukemia: clinicomolecular characteristics, response to therapy, and outcomes. Cancer. 2016;122(22):3484–3491.
- Goel S, Hall J, Pradhan K, et al. High prevalence and allele burden-independent prognostic importance of p53 mutations in an inner-city MDS/AML cohort. Leukemia. 2016;30(8):1793–1795.
- Döhner H, Dolnik A, Tang L, et al. Cytogenetics and gene mutations influence survival in older patients with acute myeloid leukemia treated with azacitidine or conventional care. Leukemia. 2018;32(12):2546–2557.
- Grob T, Al Hinai ASA, Sanders MA, et al. Molecular characterization of mutant TP53 acute myeloid leukemia and high-risk myelodysplastic syndrome. Blood. 2022;139(15):2347–2354.
- Granowicz EM, Jonas BA. Targeting TP53-mutated acute myeloid leukemia: research and clinical developments. Onco Targets Ther. 2022;15:423–436.
- Molica M, Mazzone C, Niscola P, et al. TP53 mutations in acute myeloid leukemia: still a daunting challenge? Front Oncol. 2020;10:610820.
- Klimovich B, Meyer L, Merle N. Partial p53 reactivation is sufficient to induce cancer regression. J Exp Clin Cancer Res. 2022;41(1):80.